

Mitochondrial disease in adults: what's old and what's new?
- PMID: 26612854
- PMCID: PMC4693502
- DOI: 10.15252/emmm.201505079
Mitochondrial disease in adults: what's old and what's new?
Abstract
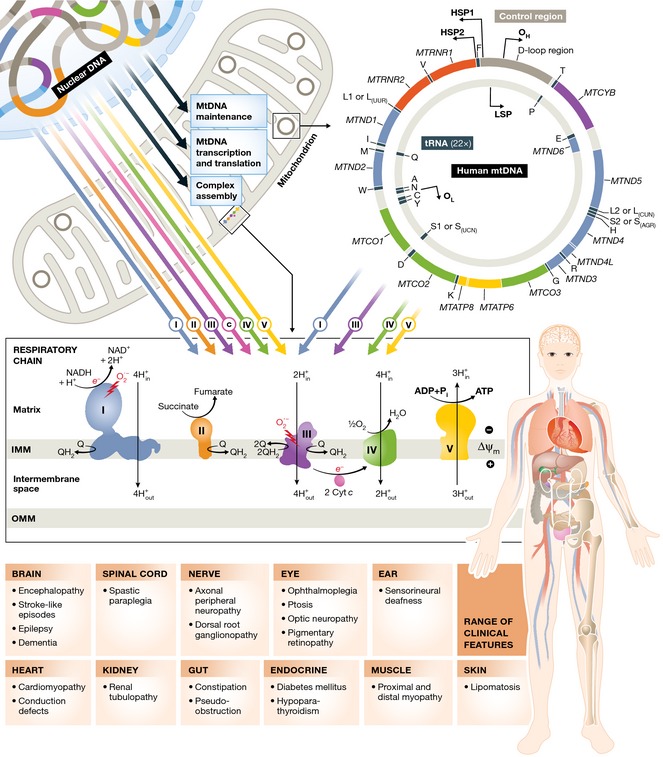
Ten years ago, there was an emerging view that the molecular basis for adult mitochondrial disorders was largely known and that the clinical phenotypes had been well described. Nothing could have been further from the truth. The establishment of large cohorts of patients has revealed new aspects of the clinical presentation that were not previously appreciated. Over time, this approach is starting to provide an accurate understanding of the natural history of mitochondrial disease in adults. Advances in molecular diagnostics, underpinned by next generation sequencing technology, have identified novel molecular mechanisms. Recently described mitochondrial disease phenotypes have disparate causes, and yet share common mechanistic themes. In particular, disorders of mtDNA maintenance have emerged as a major cause of mitochondrial disease in adults. Progressive mtDNA depletion and the accumulation of mtDNA mutations explain some of the clinical features, but the genetic and cellular processes responsible for the mtDNA abnormalities are not entirely clear in each instance. Unfortunately, apart from a few specific examples, treatments for adult mitochondrial disease have not been forthcoming. However, the establishment of international consortia, and the first multinational randomised controlled trial, have paved the way for major progress in the near future, underpinned by growing interest from the pharmaceutical industry. Adult mitochondrial medicine is, therefore, in its infancy, and the challenge is to harness the new understanding of its molecular and cellular basis to develop treatments of real benefit to patients.
Keywords: mitochondrial DNA; mitochondrial disease; mitochondrial encephalomyopathy; myopathy; neurometabolic.
© 2015 The Author. Published under the terms of the CC BY 4.0 license.
Figures
References
-
- Alavi MV, Fuhrmann N, Nguyen HP, Yu‐Wai‐Man P, Heiduschka P, Chinnery PF, Wissinger B (2009) Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp Neurol 220: 404–409 - PubMed
-
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo‐Kottler B, Auburger G et al (2000) OPA1, encoding a dynamin‐related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215 - PubMed
-
- Amati‐Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R et al (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 131: 338–351 - PubMed
-
- Baruffini E, Dallabona C, Invernizzi F, Yarham JW, Melchionda L, Blakely EL, Lamantea E, Donnini C, Santra S, Vijayaraghavan S et al (2013) MTO1 mutations are associated with hypertrophic cardiomyopathy and lactic acidosis and cause respiratory chain deficiency in humans and yeast. Hum Mutat 34: 1501–1509 - PMC - PubMed
-
- Bates MG, Hollingsworth KG, Newman JH, Jakovljevic DG, Blamire AM, Macgowan GA, Keavney BD, Chinnery PF, Turnbull DM, Taylor RW et al (2013) Concentric hypertrophic remodelling and subendocardial dysfunction in mitochondrial DNA point mutation carriers. Eur Heart J Cardiovasc Imaging 14: 650–658 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
