

Assessing the Unseen Bacterial Diversity in Microbial Communities
- PMID: 26615218
- PMCID: PMC4700968
- DOI: 10.1093/gbe/evv234
Assessing the Unseen Bacterial Diversity in Microbial Communities
Abstract
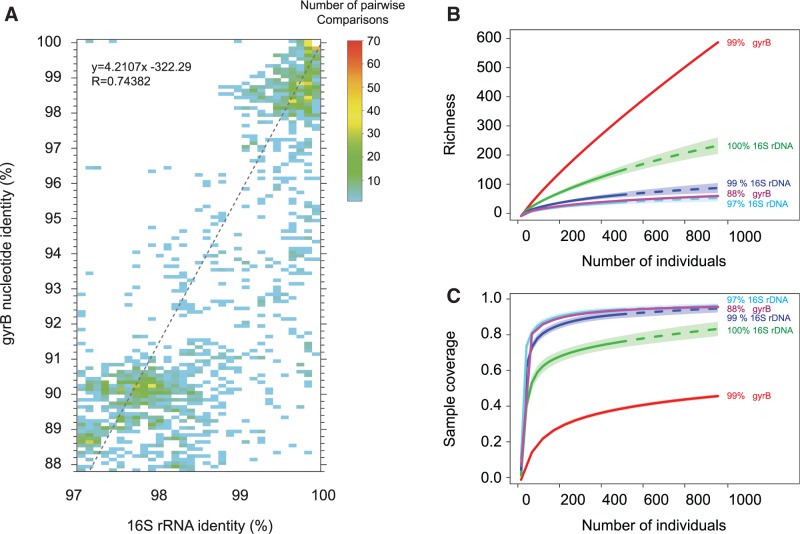
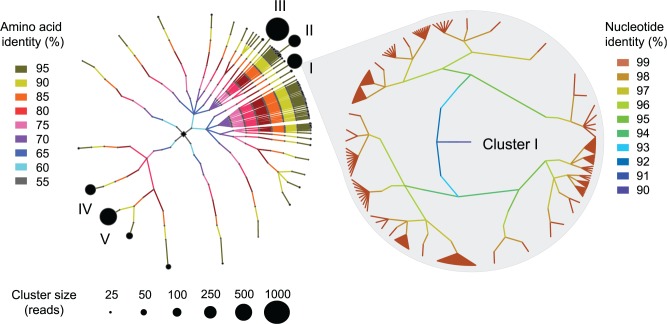
For both historical and technical reasons, 16S ribosomal RNA has been the most common molecular marker used to analyze the contents of microbial communities. However, its slow rate of evolution hinders the resolution of closely related bacteria--individual 16S-phylotypes, particularly when clustered at 97% sequence identity, conceal vast amounts of species- and strain-level variation. Protein-coding genes, which evolve more quickly, are useful for differentiating among more recently diverged lineages, but their application is complicated by difficulties in designing low-redundancy primers that amplify homologous regions from distantly related taxa. Given the now-common practice of multiplexing hundreds of samples, adopting new genes usually entails the synthesis of large sets of barcoded primers. To circumvent problems associated with use of protein-coding genes to survey microbial communities, we develop an approach--termed phyloTAGs--that offers an automatic solution for primer design and can be easily adapted to target different taxonomic groups and/or different protein-coding regions. We applied this method to analyze diversity within the gorilla gut microbiome and recovered hundreds of strains that went undetected after deep-sequencing of 16S rDNA amplicons. PhyloTAGs provides a powerful way to recover the fine-level diversity within microbial communities and to study stability and dynamics of bacterial populations.
Keywords: bacterial species; bacterial strain diversity; community profiling; microbiome; phylotypes; population structure.
© The Author(s) 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Brettar I, Moore ER, Hofle MG. 2001. Phylogeny and abundance of novel denitrifying bacteria isolated from the water column of the central Baltic Sea. Microb Ecol 42:295–305. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
