

Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity
- PMID: 26619011
- PMCID: PMC4744099
- DOI: 10.1038/nbt.3391
Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity
Abstract
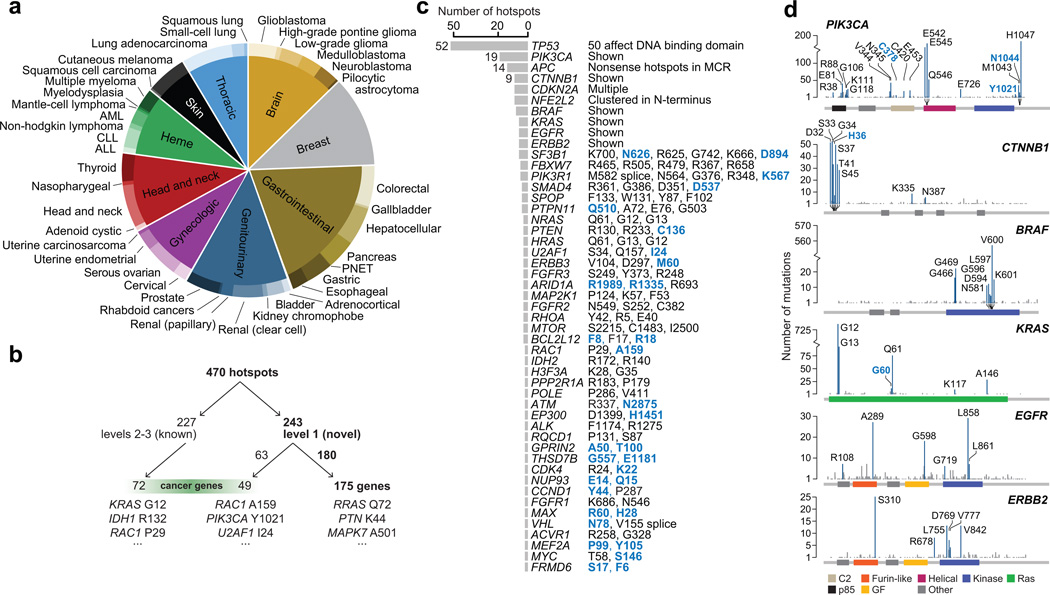
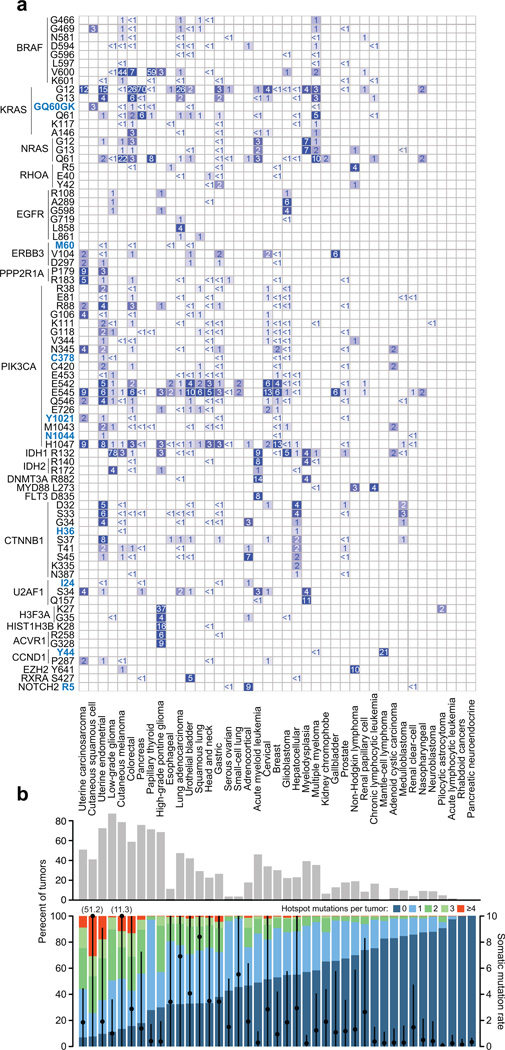
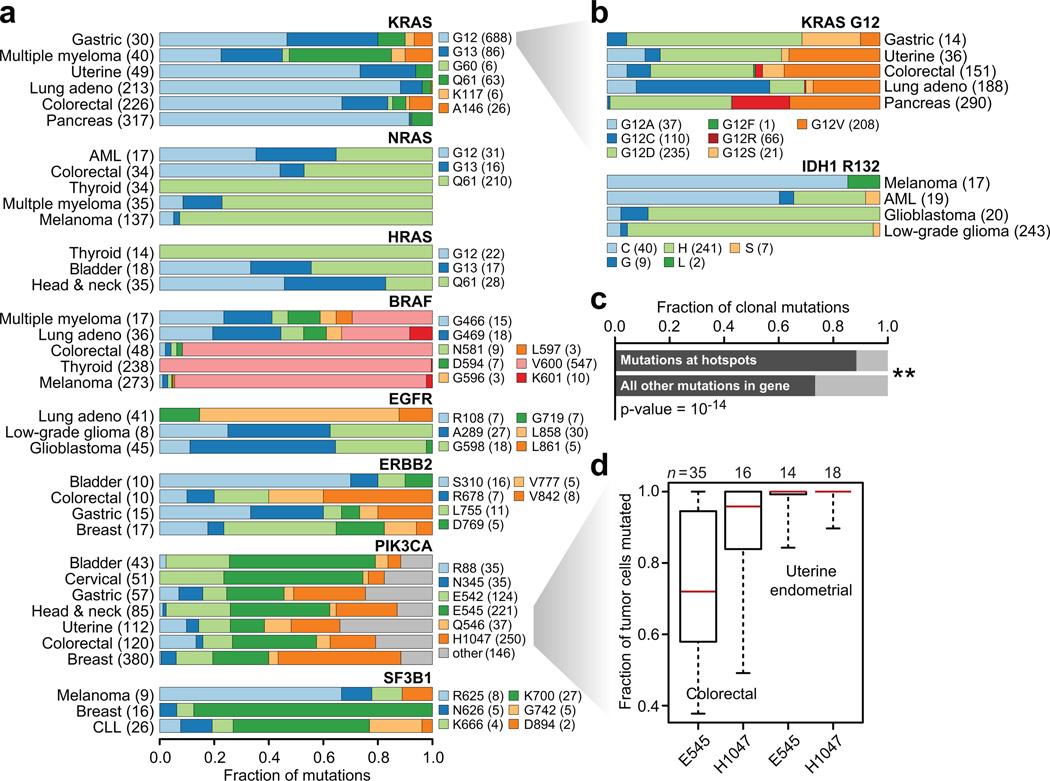
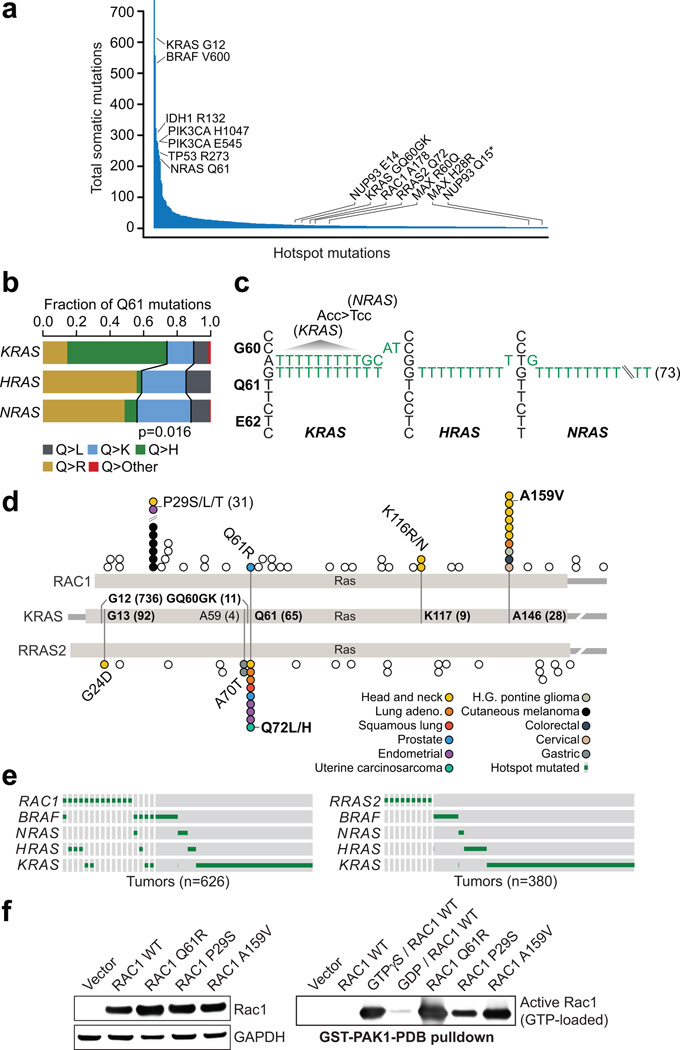
Mutational hotspots indicate selective pressure across a population of tumor samples, but their prevalence within and across cancer types is incompletely characterized. An approach to detect significantly mutated residues, rather than methods that identify recurrently mutated genes, may uncover new biologically and therapeutically relevant driver mutations. Here, we developed a statistical algorithm to identify recurrently mutated residues in tumor samples. We applied the algorithm to 11,119 human tumors, spanning 41 cancer types, and identified 470 somatic substitution hotspots in 275 genes. We find that half of all human tumors possess one or more mutational hotspots with widespread lineage-, position- and mutant allele-specific differences, many of which are likely functional. In total, 243 hotspots were novel and appeared to affect a broad spectrum of molecular function, including hotspots at paralogous residues of Ras-related small GTPases RAC1 and RRAS2. Redefining hotspots at mutant amino acid resolution will help elucidate the allele-specific differences in their function and could have important therapeutic implications.
Figures
References
-
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. - PubMed
Online Methods-only References
-
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals of Statistics. 2001;29:1165–1188.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
