

doi: 10.1186/s13059-015-0831-x.
HiC-Pro: an optimized and flexible pipeline for Hi-C data processing
Affiliations
- PMID: 26619908
- PMCID: PMC4665391
- DOI: 10.1186/s13059-015-0831-x
Item in Clipboard
HiC-Pro: an optimized and flexible pipeline for Hi-C data processing
Genome Biol.
.
Abstract
HiC-Pro is an optimized and flexible pipeline for processing Hi-C data from raw reads to normalized contact maps. HiC-Pro maps reads, detects valid ligation products, performs quality controls and generates intra- and inter-chromosomal contact maps. It includes a fast implementation of the iterative correction method and is based on a memory-efficient data format for Hi-C contact maps. In addition, HiC-Pro can use phased genotype data to build allele-specific contact maps. We applied HiC-Pro to different Hi-C datasets, demonstrating its ability to easily process large data in a reasonable time. Source code and documentation are available at http://github.com/nservant/HiC-Pro .
Figures
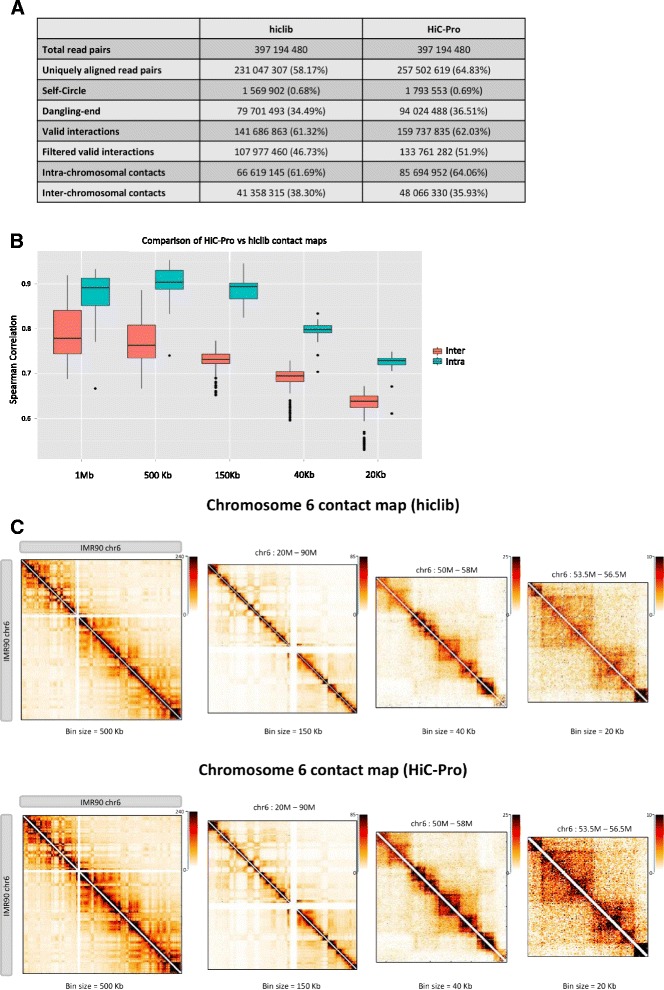
Comparison of HiC-Pro and hiclib processing. a Both pipelines generate concordant results across processing steps. The fraction of uniquely aligned read pairs is calculated on the total number of initial reads. Self-circle and dangling-end fractions are calculated on the total number of aligned read pairs. Intra- and inter-chromosomal contacts are calculated as a fraction of filtered valid interactions. b Boxplots of the Spearman correlation coefficients of intra- and inter-chromosomal maps generated at different resolutions by both pipelines. c Chromosome 6 contact maps generated by hiclib (top) and HiC-Pro (bottom) at different resolutions. The chromatin interaction data generated by the two pipelines are highly similar
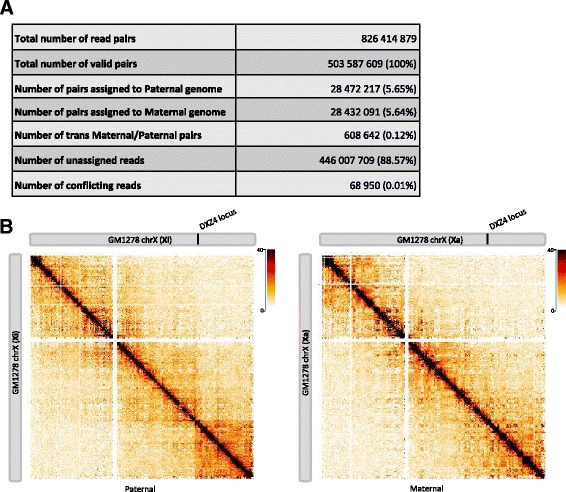
Allele-specific analysis. a Allele-specific analysis of the GM12878 cell line. Phasing data were gathered from the Illumina Platinum Genomes Project. In total, 2,239,492 high quality SNPs from GM12878 data were used to distinguish both alleles. Around 6 % of the read pairs were assigned to each parental allele and used to build the allele-specific contact maps. b Intra-chromosomal contact maps of inactive and active X chromosome of the GM12878 cell line at 500-kb resolution. The inactive copy of chromosome X is partitioned into two mega-domains which are not seen in the active X chromosome. The boundary between the two mega-domains lies near the DXZ4 micro-satellite
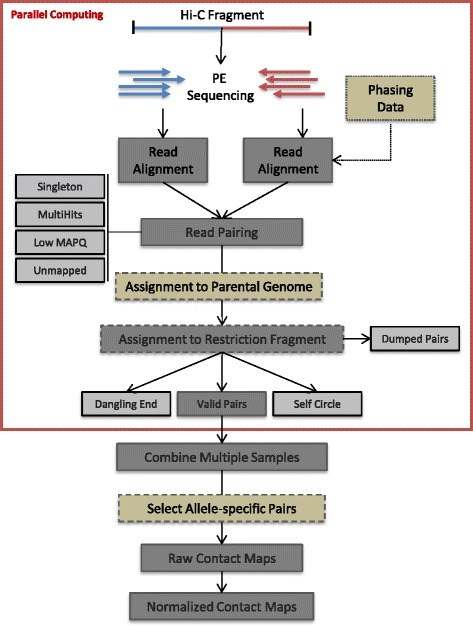
HiC-Pro workflow. Reads are first aligned on the reference genome. Only uniquely aligned reads are kept and assigned to a restriction fragment. Interactions are then classified and invalid pairs are discarded. If phased genotyping data and N-masked genome are provided, HiC-Pro will align the reads and assign them to a parental genome. For the Hi-C protocol based on restriction enzyme digestion, the read pairs will then be assigned to a restriction fragment and invalid ligation products will be filtered out. These first steps can be performed in parallel for each read chunk. Data from multiple chunks are then merged and binned to generate a single genome-wide interaction map. For allele-specific analysis, only pairs with at least one allele-specific read are used to build the contact maps. The normalization is finally applied to remove Hi-C systematic bias on the genome-wide contact map. MAPQ Mapping Quality , PE paired end
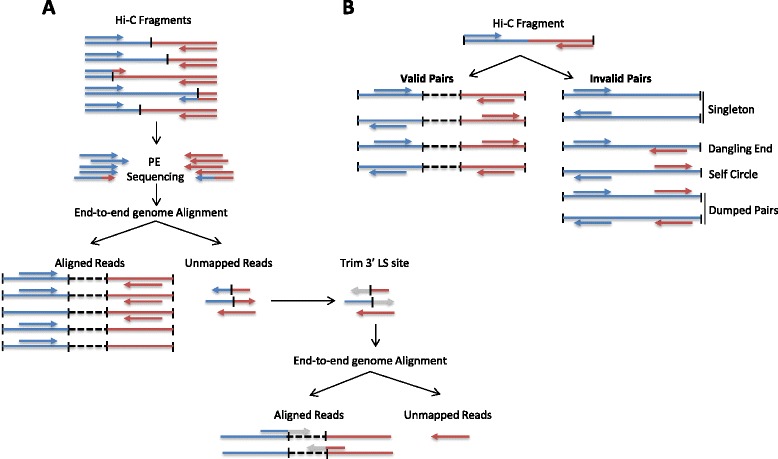
Read pair alignment and filtering. a Read pairs are first independently aligned to the reference genome using an end-to-end algorithm. Then, reads spanning the ligation junction which were not aligned in the first step are trimmed at the ligation site and their 5′ extremity is realigned on the genome. All aligned reads after these two steps are used for further analysis. b According to the Hi-C protocol, digested fragments are ligated together to generate Hi-C products. A valid Hi-C product is expected to involve two different restriction fragments. Read pairs aligned on the same restriction fragment are classified as dangling end or self-circle products, and are not used to generate the contact maps. PE paired end, LS Ligation Site
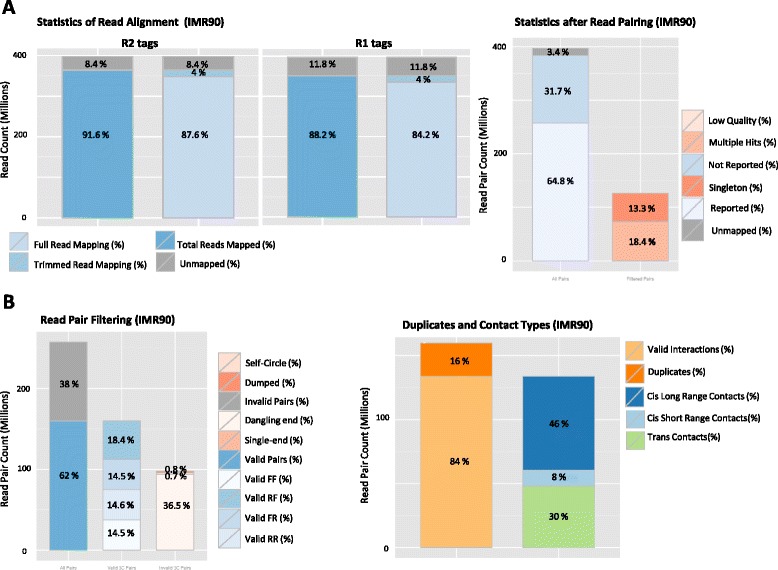
HiC-Pro quality controls. Quality controls reported by HiC-Pro (IMR90, Dixon et al. data). a Quality control on read alignment and pairing. Low quality alignment, singleton and multiple hits are usually removed at this step. b Read pair filtering. Read pairs are assigned to a restriction fragment. Invalid pairs, such as dangling-end and self-circle, are good indicators of the library quality and are tracked but discarded for subsequent further analysis. The fractions of duplicated reads, as well as short range versus long range interactions, are also reported
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
