

Neofunction of ACVR1 in fibrodysplasia ossificans progressiva
- PMID: 26621707
- PMCID: PMC4687587
- DOI: 10.1073/pnas.1510540112
Neofunction of ACVR1 in fibrodysplasia ossificans progressiva
Abstract
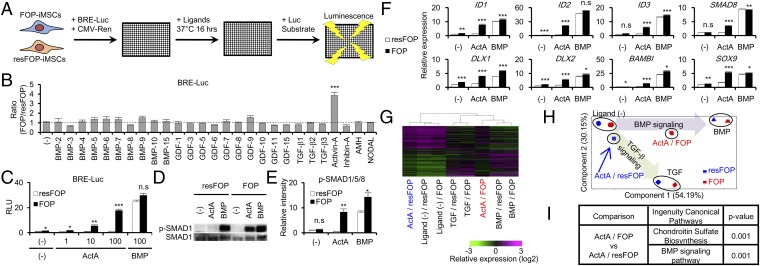
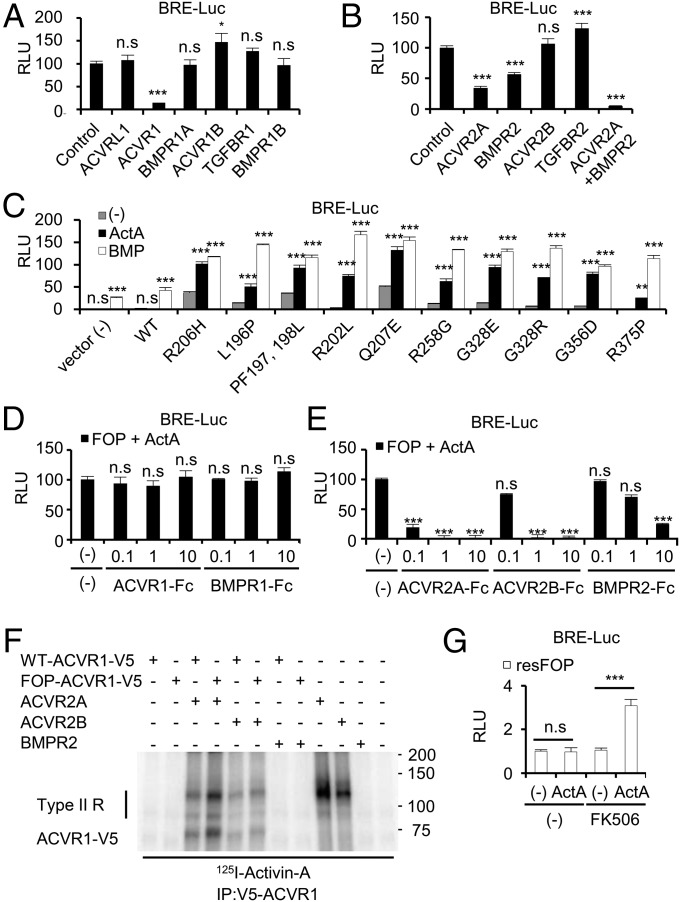
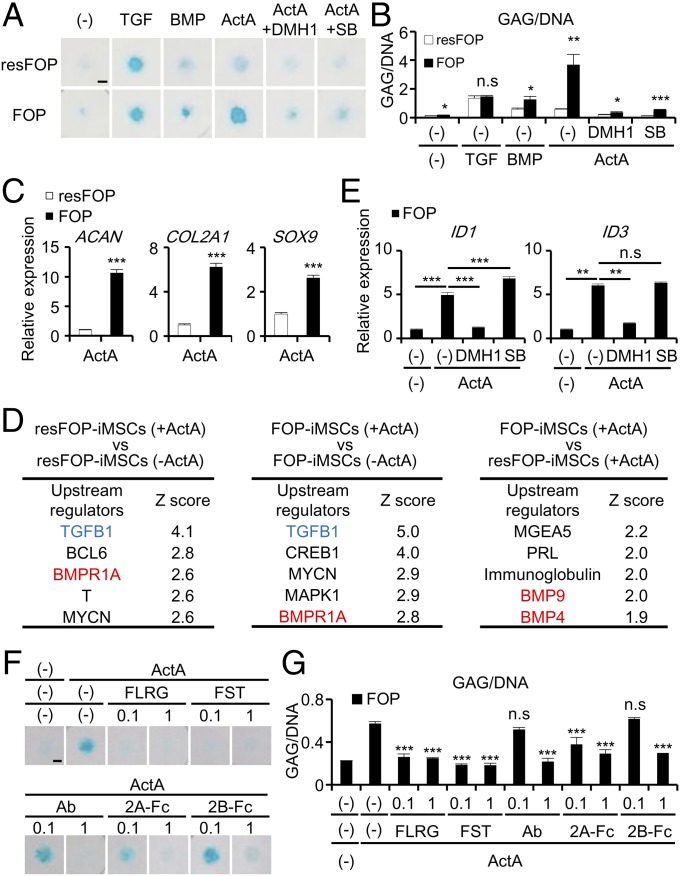
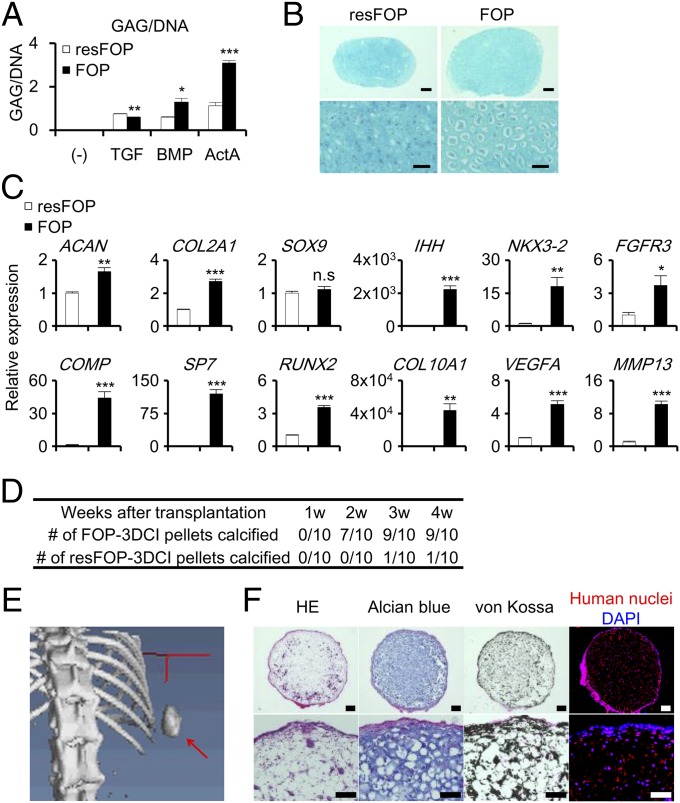
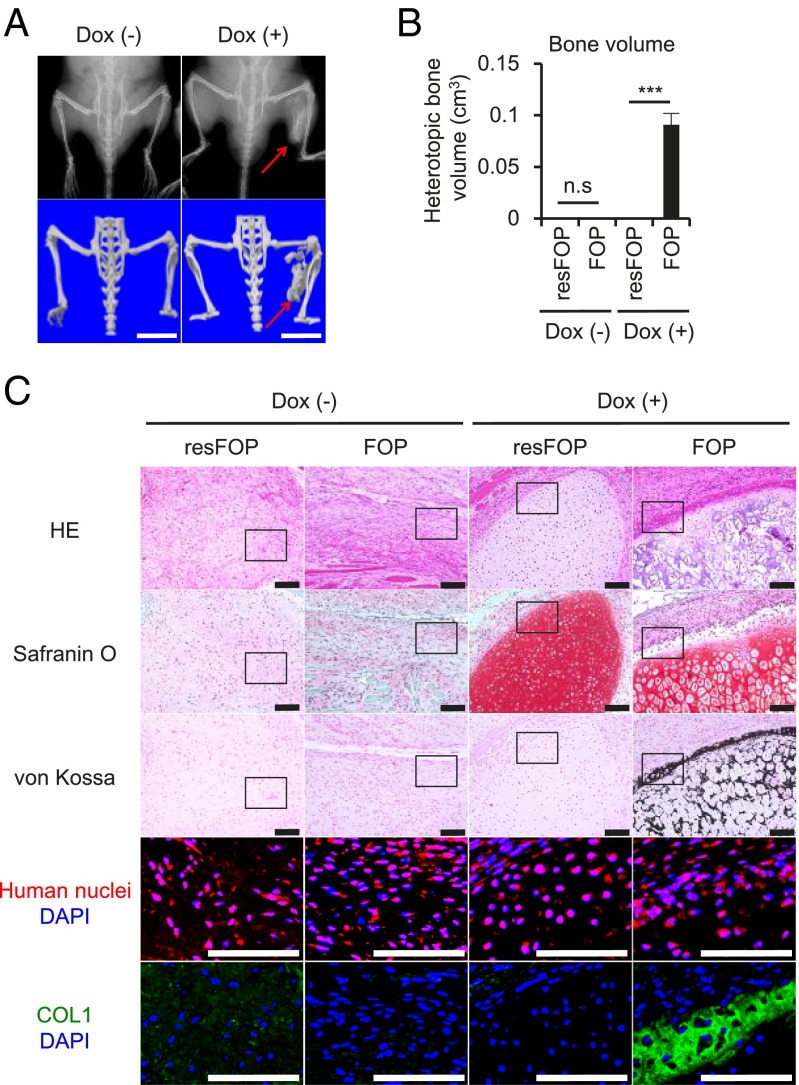
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disease characterized by extraskeletal bone formation through endochondral ossification. FOP patients harbor point mutations in ACVR1 (also known as ALK2), a type I receptor for bone morphogenetic protein (BMP). Two mechanisms of mutated ACVR1 (FOP-ACVR1) have been proposed: ligand-independent constitutive activity and ligand-dependent hyperactivity in BMP signaling. Here, by using FOP patient-derived induced pluripotent stem cells (FOP-iPSCs), we report a third mechanism, where FOP-ACVR1 abnormally transduces BMP signaling in response to Activin-A, a molecule that normally transduces TGF-β signaling but not BMP signaling. Activin-A enhanced the chondrogenesis of induced mesenchymal stromal cells derived from FOP-iPSCs (FOP-iMSCs) via aberrant activation of BMP signaling in addition to the normal activation of TGF-β signaling in vitro, and induced endochondral ossification of FOP-iMSCs in vivo. These results uncover a novel mechanism of extraskeletal bone formation in FOP and provide a potential new therapeutic strategy for FOP.
Keywords: BMP; TGF; fibrodysplasia ossificans progressiva; heterotopic ossification; iPSC.
Conflict of interest statement
Conflict of interest statement: K. Hino, K. Horigome, and H.E. are employees of Sumitomo Dainippon Pharma Co., Ltd; and M.I. and J.T. are supported by a research fund from Sumitomo Dainippon Pharma Co., Ltd.
Figures
References
-
- Kaplan F, et al. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3(3-4):183–188.
-
- Shore E, Feldman G, Xu M, Kaplan F. The genetics of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3(3-4):201–204.
-
- Kaplan FS, Groppe J, Pignolo RJ, Shore EM. Morphogen receptor genes and metamorphogenes: Skeleton keys to metamorphosis. Ann N Y Acad Sci. 2007;1116:113–133. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
