

Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1α-dependent manner
- PMID: 26621917
- PMCID: PMC4727419
- DOI: 10.1194/jlr.M063412
Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1α-dependent manner
Abstract
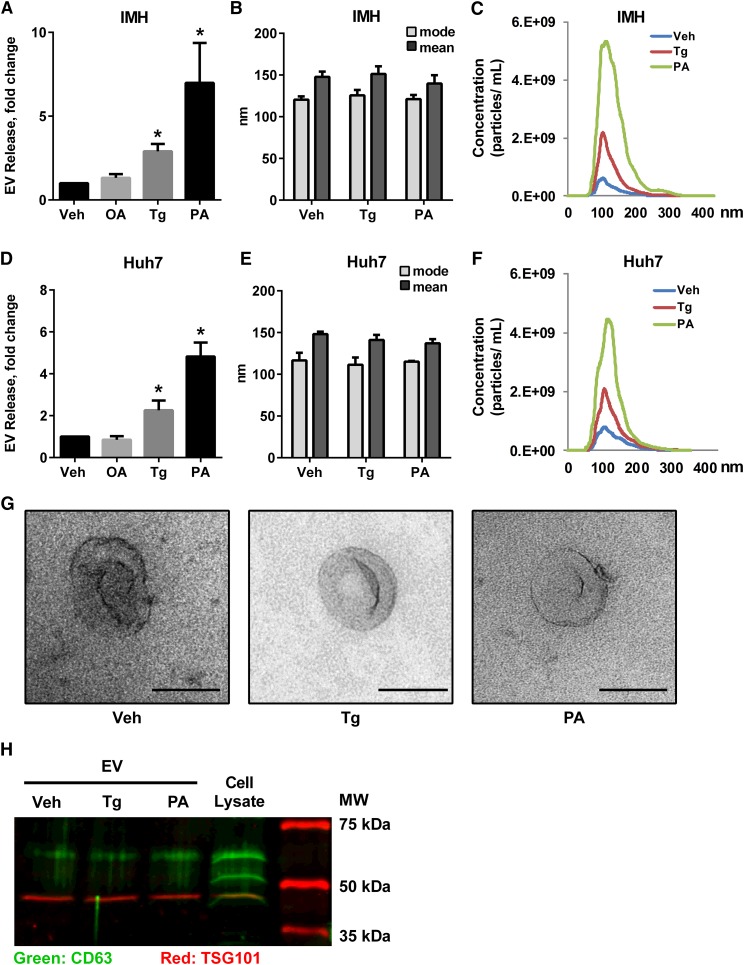
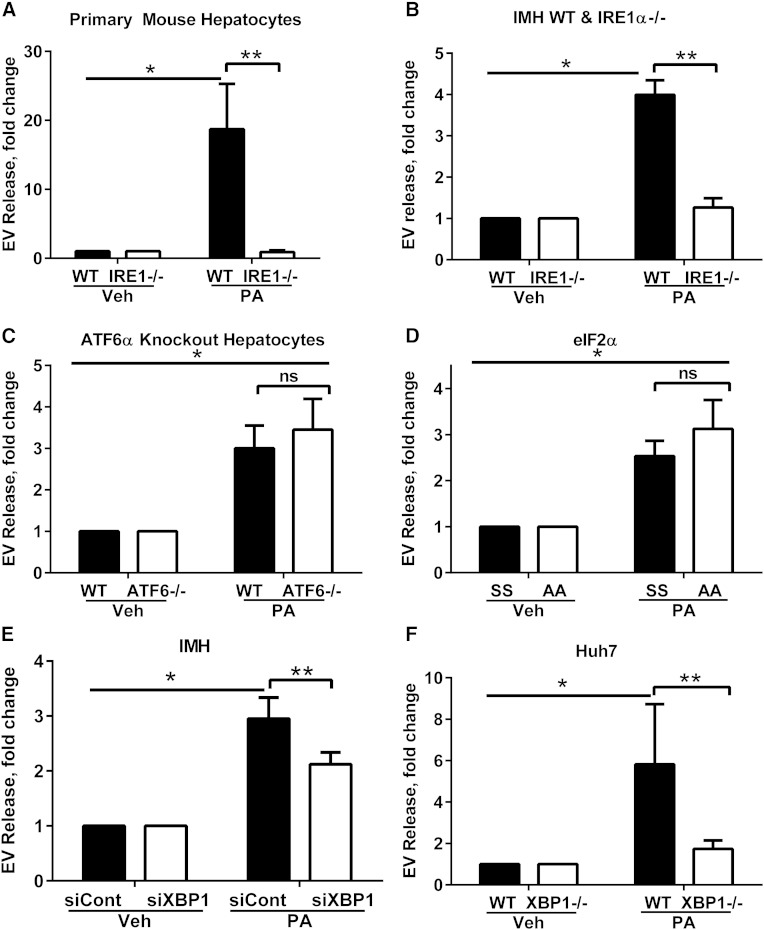
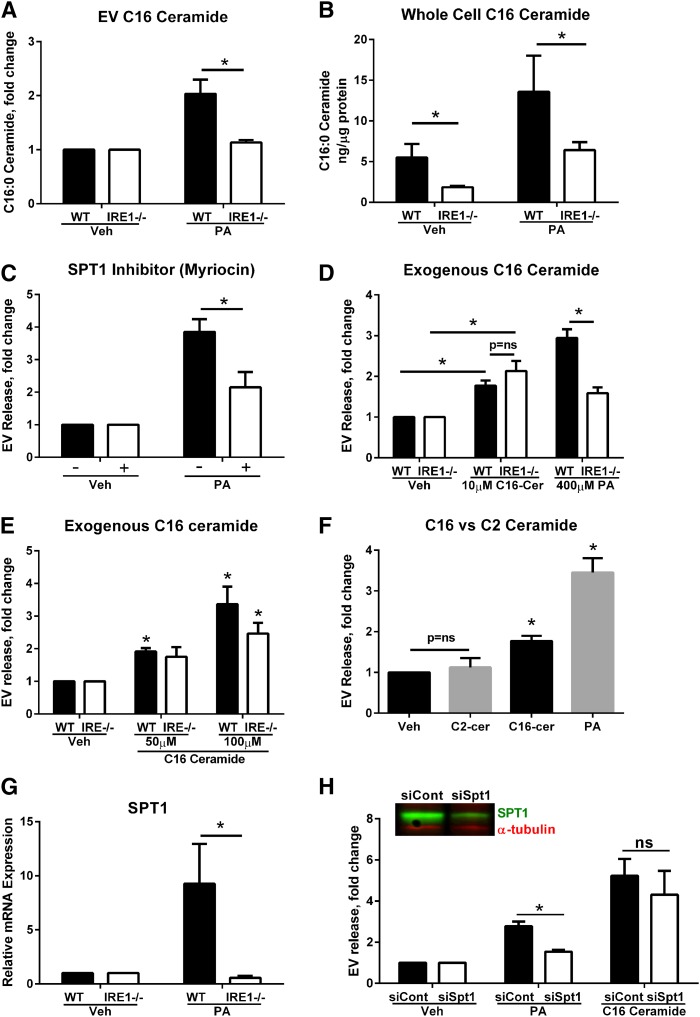
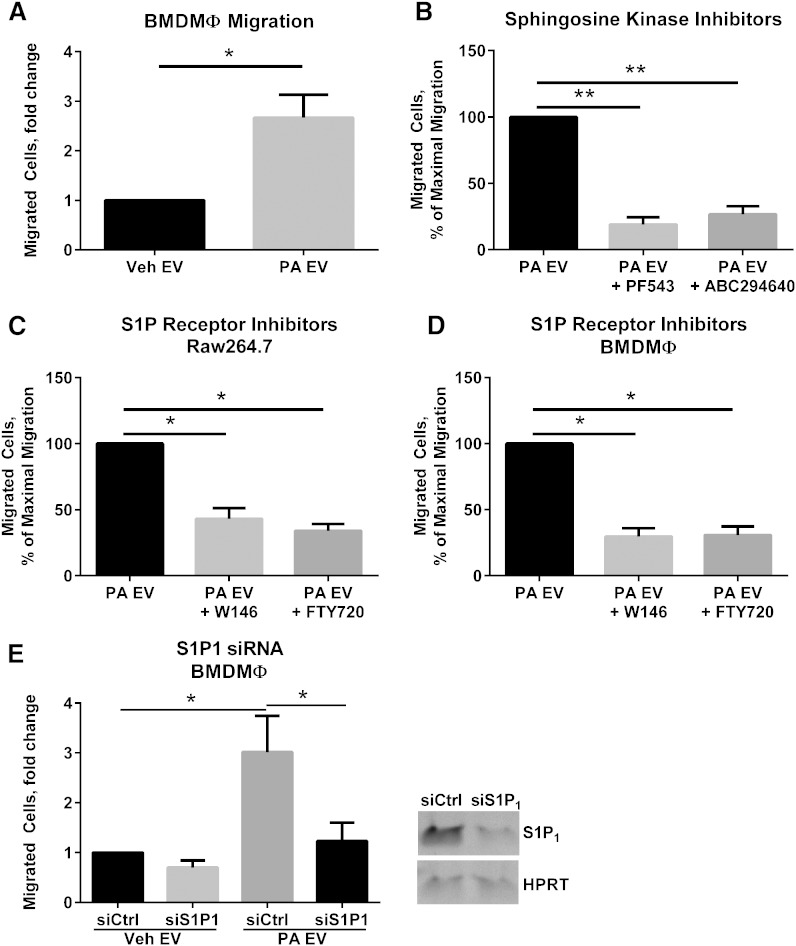
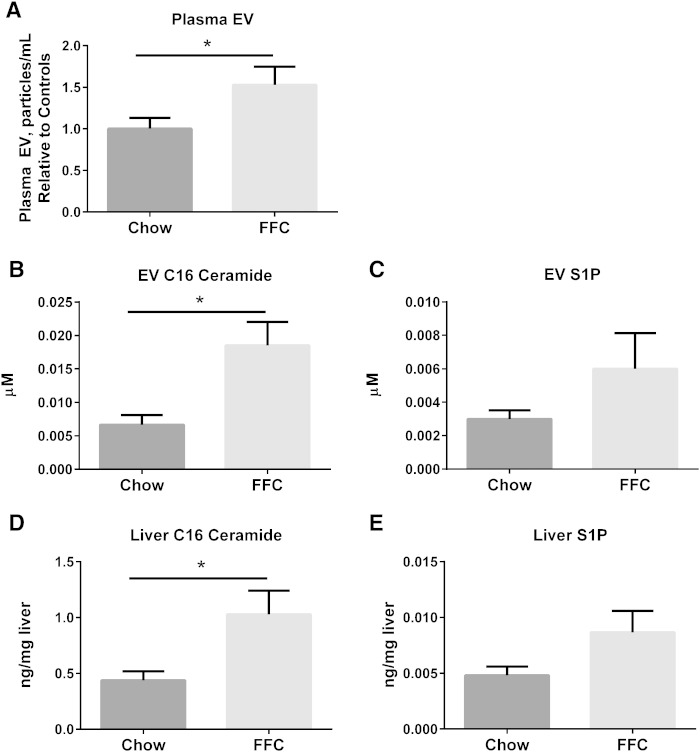
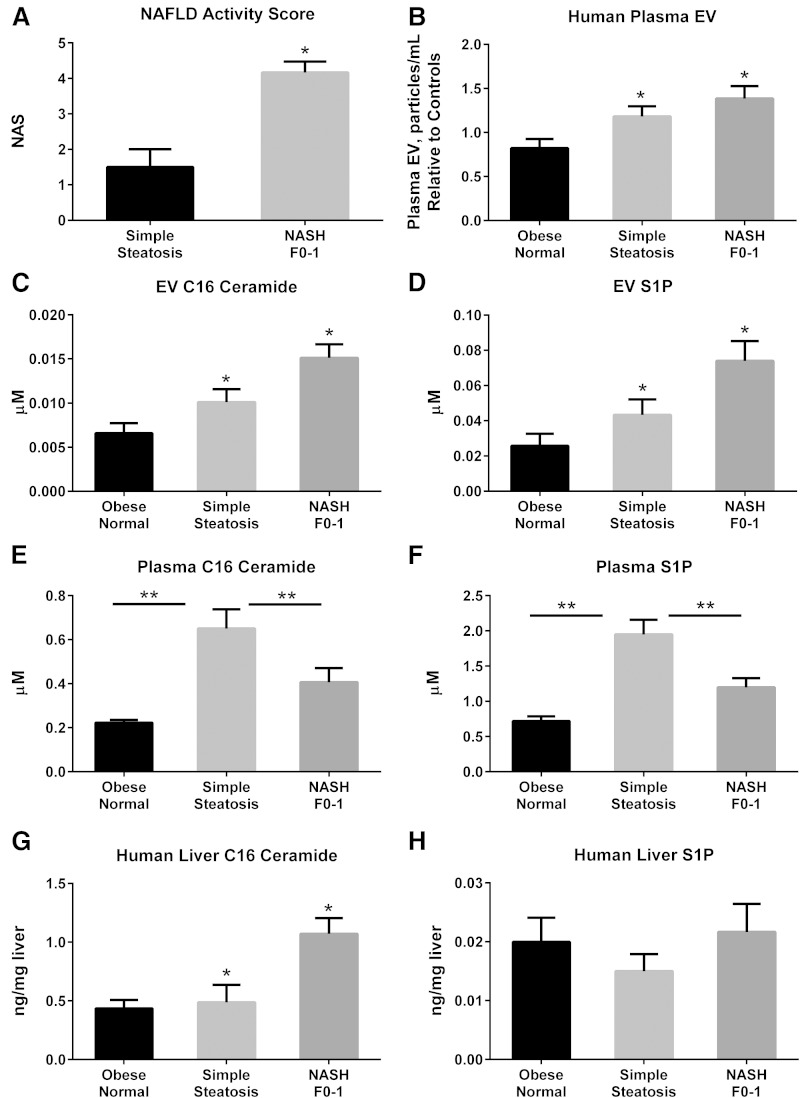
Nonalcoholic steatohepatitis (NASH) is a lipotoxic disease wherein activation of endoplasmic reticulum (ER) stress response and macrophage-mediated hepatic inflammation are key pathogenic features. However, the lipid mediators linking these two observations remain elusive. We postulated that ER stress-regulated release of pro-inflammatory extracellular vesicles (EVs) from lipotoxic hepatocytes may be this link. EVs were isolated from cell culture supernatants of hepatocytes treated with palmitate (PA) to induce lipotoxic ER stress, characterized by immunofluorescence, Western blotting, electron microscopy, and nanoparticle tracking analysis. Sphingolipids were measured by tandem mass spectrometry. EVs were employed in macrophage chemotaxis assays. PA induced significant EV release. Because PA activates ER stress, we used KO hepatocytes to demonstrate that PA-induced EV release was mediated by inositol requiring enzyme 1α (IRE1α)/X-box binding protein-1. PA-induced EVs were enriched in C16:0 ceramide in an IRE1α-dependent manner, and activated macrophage chemotaxis via formation of sphingosine-1-phosphate (S1P) from C16:0 ceramide. This chemotaxis was blocked by sphingosine kinase inhibitors and S1P receptor inhibitors. Lastly, elevated circulating EVs in experimental and human NASH demonstrated increased C16:0 ceramide. PA induces C16:0 ceramide-enriched EV release in an IRE1α-dependent manner. The ceramide metabolite, S1P, activates macrophage chemotaxis, a potential mechanism for the recruitment of macrophages to the liver under lipotoxic conditions.
Keywords: endoplasmic reticulum stress; exosome; inositol requiring enzyme 1α; lipoinflammation; microvesicle; nonalcoholic steatohepatitis.
Copyright © 2016 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
Comment in
-
Extracellular vesicles and ceramide: new mediators for macrophage chemotaxis?J Lipid Res. 2016 Feb;57(2):157-8. doi: 10.1194/jlr.C066191. Epub 2016 Jan 4. J Lipid Res. 2016. PMID: 26729816 Free PMC article. No abstract available.
References
-
- Rinella M. E. 2015. Nonalcoholic fatty liver disease: a systematic review. JAMA. 313: 2263–2273. - PubMed
-
- Lassailly G., Caiazzo R., Buob D., Pigeyre M., Verkindt H., Labreuche J., Raverdy V., Leteurtre E., Dharancy S., Louvet A., et al. . 2015. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology. 149: 379–388. - PubMed
-
- Nehra V., Angulo P., Buchman A. L., and Lindor K. D.. 2001. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig. Dis. Sci. 46: 2347–2352. - PubMed
-
- Malhi H., Bronk S. F., Werneburg N. W., and Gores G. J.. 2006. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 281: 12093–12101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
