

PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain
- PMID: 26626480
- PMCID: PMC4712911
- DOI: 10.1016/j.molcel.2015.10.013
PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain
Abstract
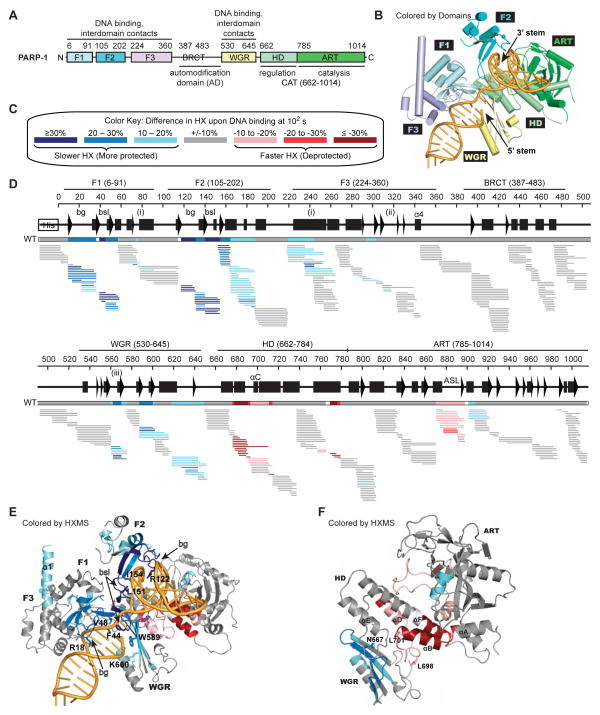
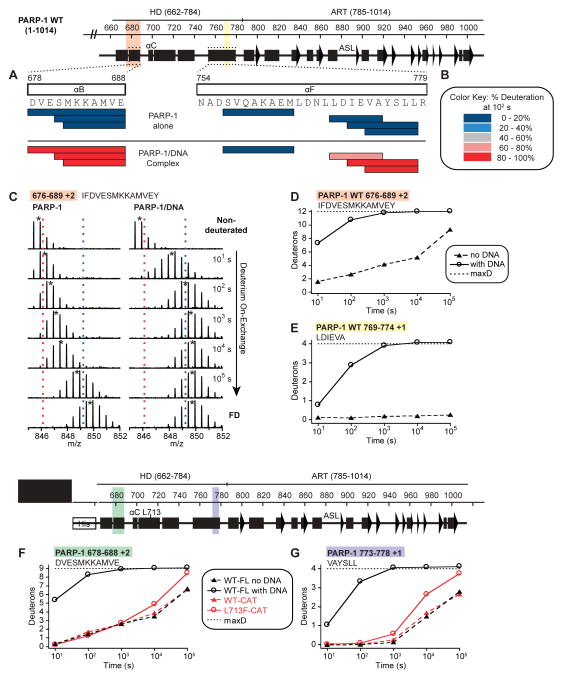
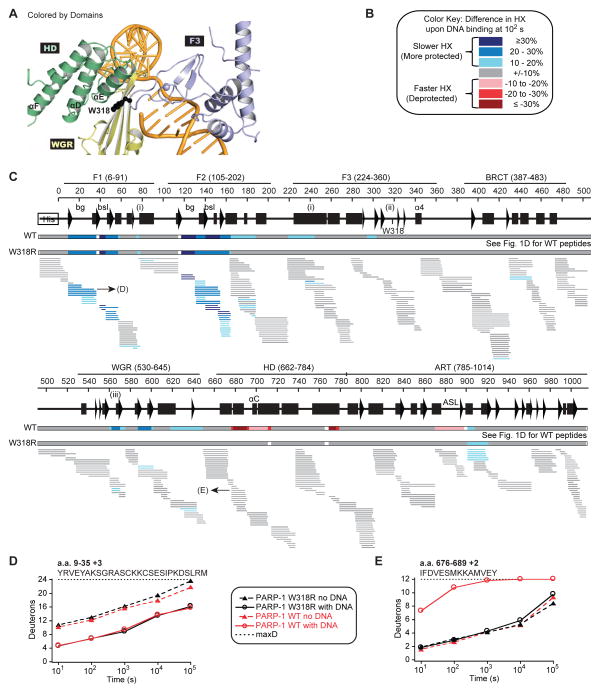
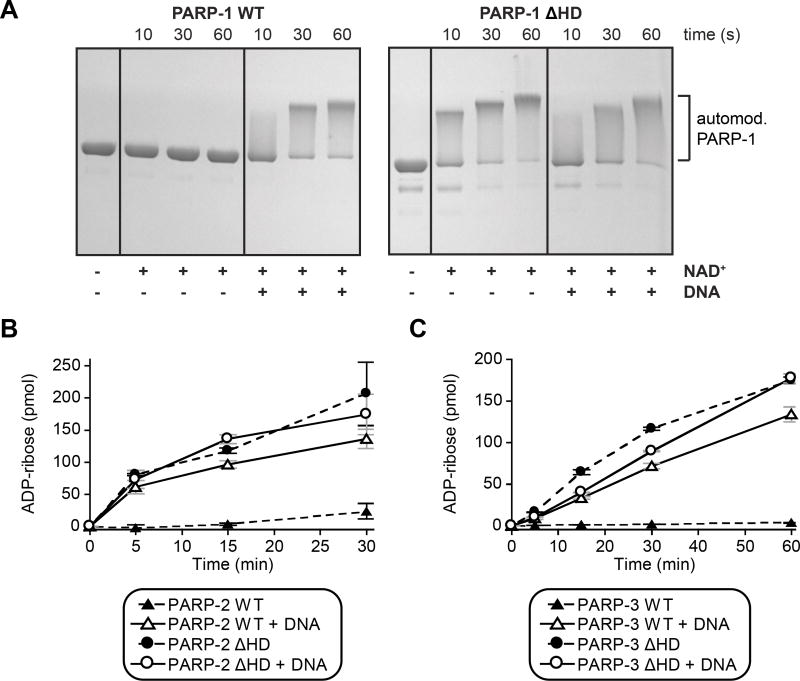
Poly(ADP-ribose) polymerase-1 (PARP-1) creates the posttranslational modification PAR from substrate NAD(+) to regulate multiple cellular processes. DNA breaks sharply elevate PARP-1 catalytic activity to mount a cell survival repair response, whereas persistent PARP-1 hyperactivation during severe genotoxic stress is associated with cell death. The mechanism for tight control of the robust catalytic potential of PARP-1 remains unclear. By monitoring PARP-1 dynamics using hydrogen/deuterium exchange-mass spectrometry (HXMS), we unexpectedly find that a specific portion of the helical subdomain (HD) of the catalytic domain rapidly unfolds when PARP-1 encounters a DNA break. Together with biochemical and crystallographic analysis of HD deletion mutants, we show that the HD is an autoinhibitory domain that blocks productive NAD(+) binding. Our molecular model explains how PARP-1 DNA damage detection leads to local unfolding of the HD that relieves autoinhibition, and has important implications for the design of PARP inhibitors.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
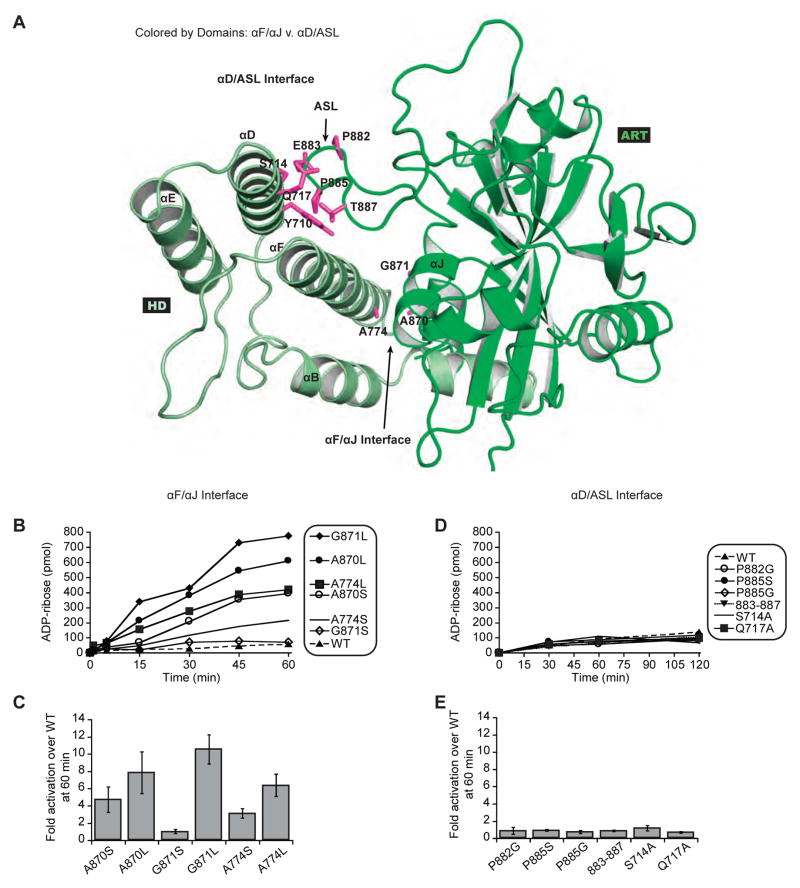
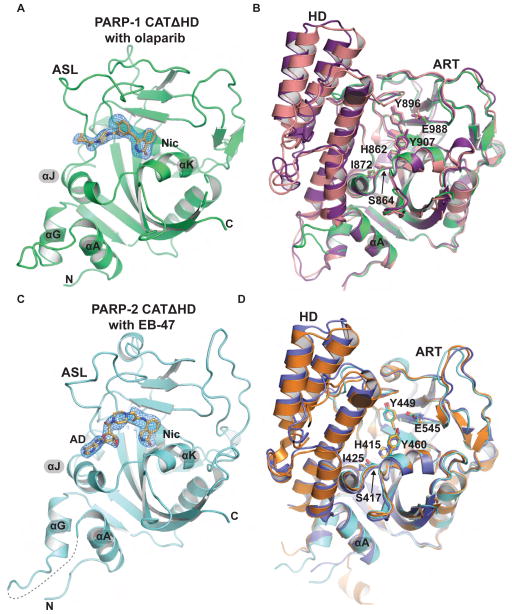
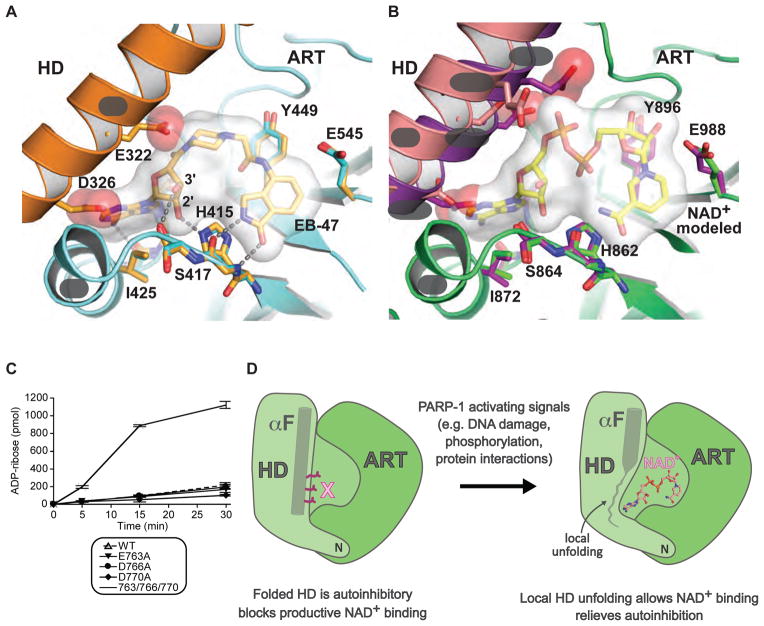
References
-
- Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
