

Structural insight into the mechanism of synergistic autoinhibition of SAD kinases
- PMID: 26626945
- PMCID: PMC4686854
- DOI: 10.1038/ncomms9953
Structural insight into the mechanism of synergistic autoinhibition of SAD kinases
Abstract
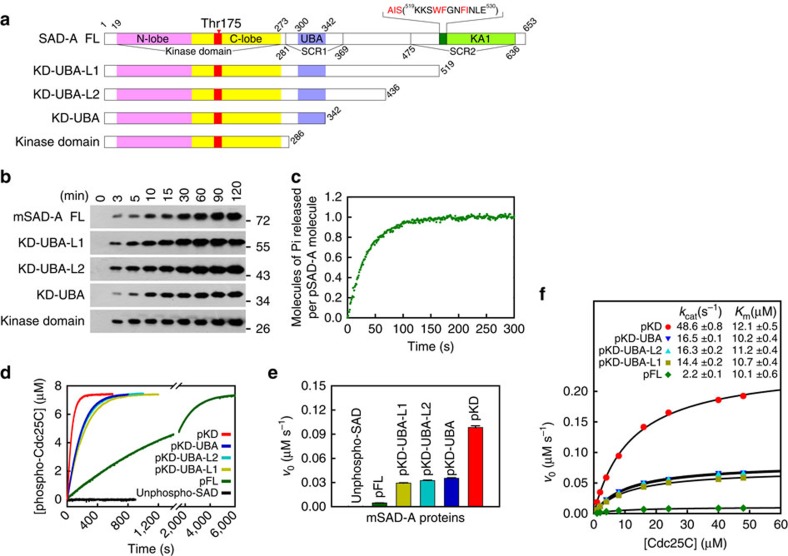
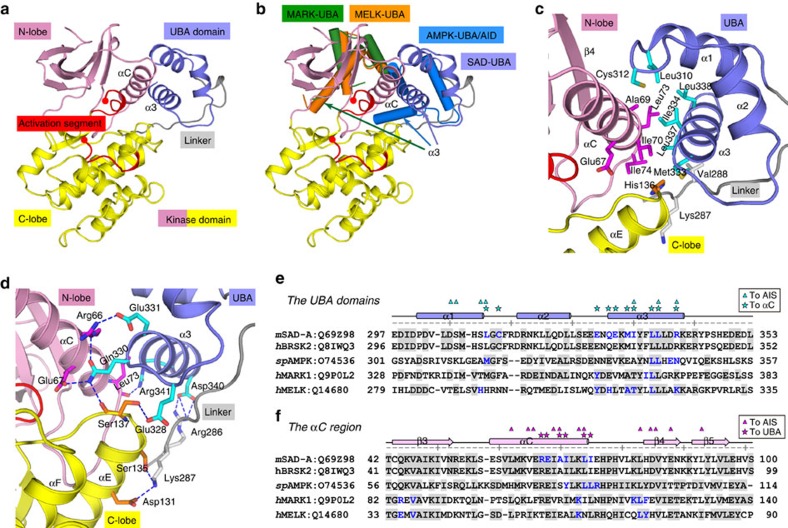
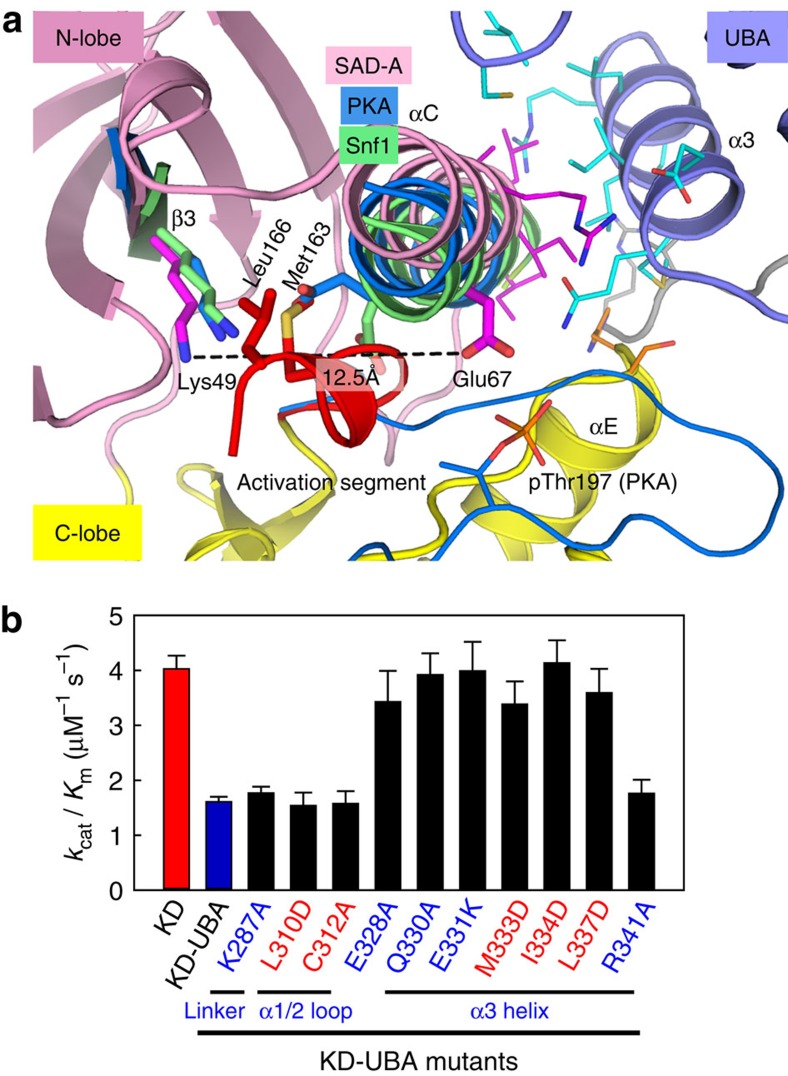
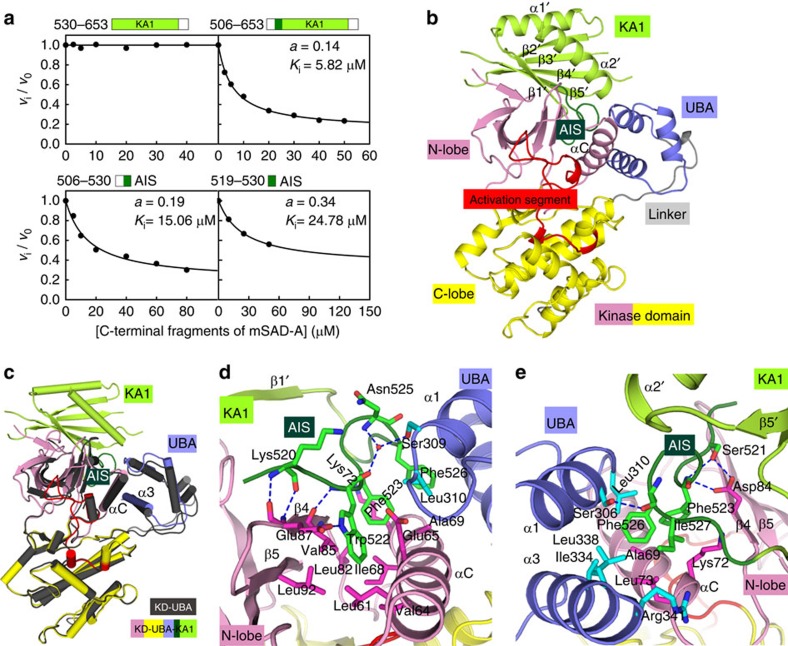
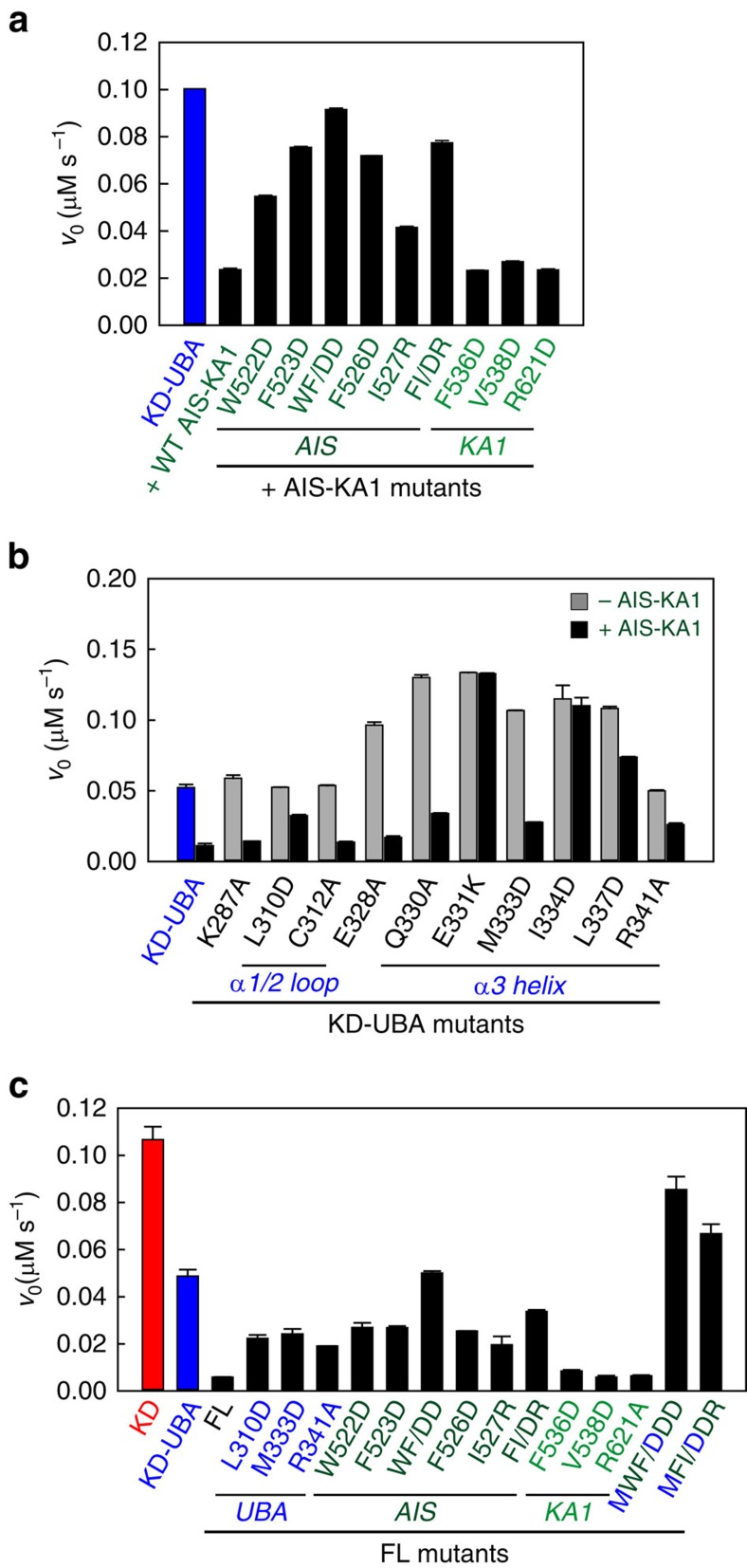
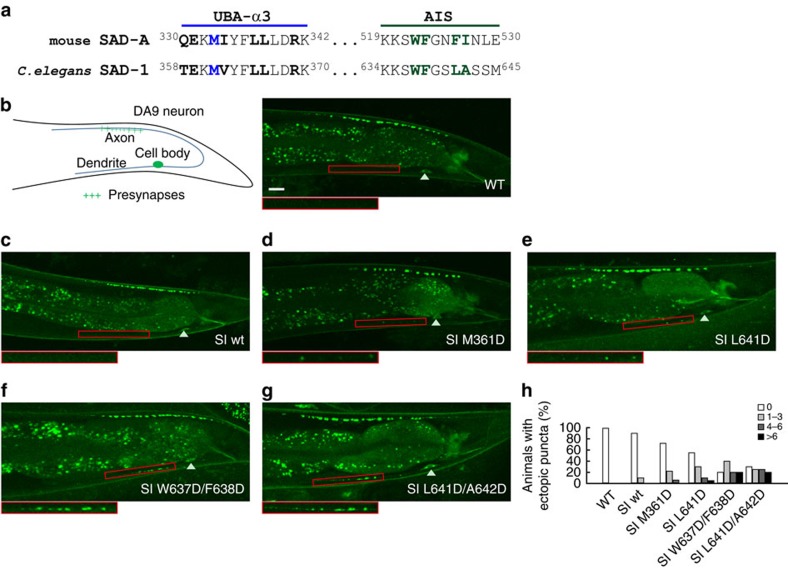
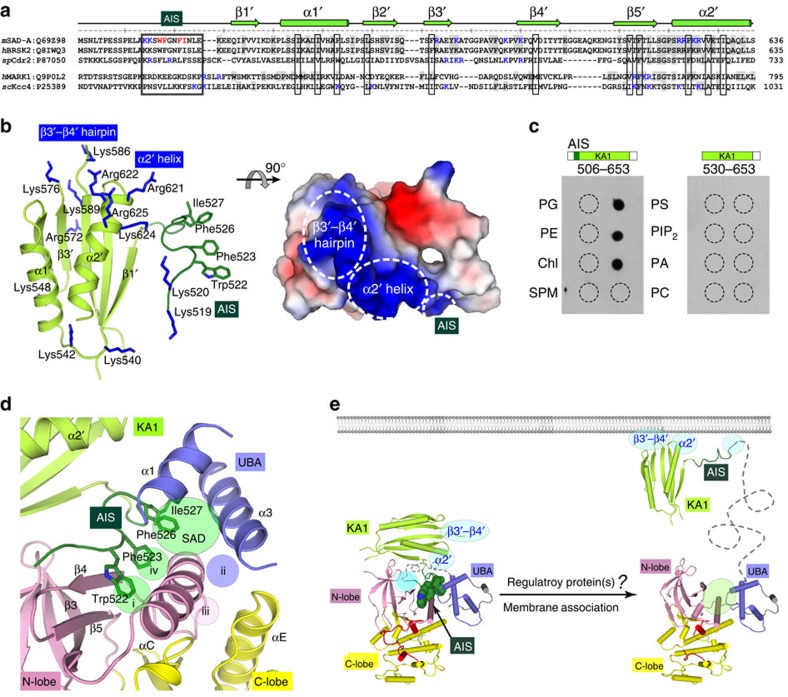
The SAD/BRSK kinases participate in various important life processes, including neural development, cell cycle and energy metabolism. Like other members of the AMPK family, SAD contains an N-terminal kinase domain followed by the characteristic UBA and KA1 domains. Here we identify a unique autoinhibitory sequence (AIS) in SAD kinases, which exerts autoregulation in cooperation with UBA. Structural studies of mouse SAD-A revealed that UBA binds to the kinase domain in a distinct mode and, more importantly, AIS nestles specifically into the KD-UBA junction. The cooperative action of AIS and UBA results in an 'αC-out' inactive kinase, which is conserved across species and essential for presynaptic vesicle clustering in C. elegans. In addition, the AIS, along with the KA1 domain, is indispensable for phospholipid binding. Taken together, these data suggest a model for synergistic autoinhibition and membrane activation of SAD kinases.
Figures
References
-
- Crump J. G., Zhen M., Jin Y. & Bargmann C. I. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron 29, 115–129 (2001). - PubMed
-
- Kishi M., Pan Y. A., Crump J. G. & Sanes J. R. Mammalian SAD kinases are required for neuronal polarization. Science 307, 929–932 (2005). - PubMed
-
- Barnes A. P. et al.. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563 (2007). - PubMed
-
- Shelly M., Cancedda L., Heilshorn S., Sumbre G. & Poo M. M. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell 129, 565–577 (2007). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
