

Zellweger spectrum disorders: clinical overview and management approach
- PMID: 26627182
- PMCID: PMC4666198
- DOI: 10.1186/s13023-015-0368-9
Zellweger spectrum disorders: clinical overview and management approach
Abstract
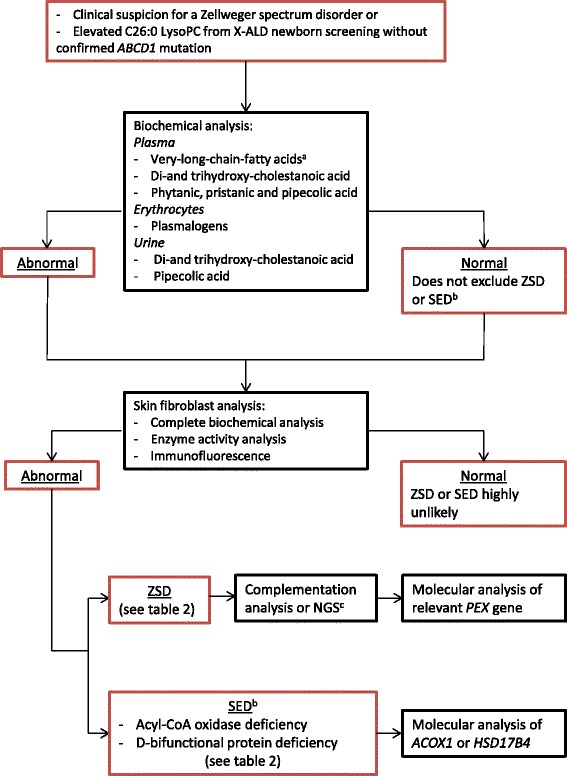
Zellweger spectrum disorders (ZSDs) represent the major subgroup within the peroxisomal biogenesis disorders caused by defects in PEX genes. The Zellweger spectrum is a clinical and biochemical continuum which can roughly be divided into three clinical phenotypes. Patients can present in the neonatal period with severe symptoms or later in life during adolescence or adulthood with only minor features. A defect of functional peroxisomes results in several metabolic abnormalities, which in most cases can be detected in blood and urine. There is currently no curative therapy, but supportive care is available. This review focuses on the management of patients with a ZSD and provides recommendations for supportive therapeutic options for all those involved in the care for ZSD patients.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
