

Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress
- PMID: 26627253
- PMCID: PMC4672794
- DOI: 10.1073/pnas.1515733112
Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress
Abstract
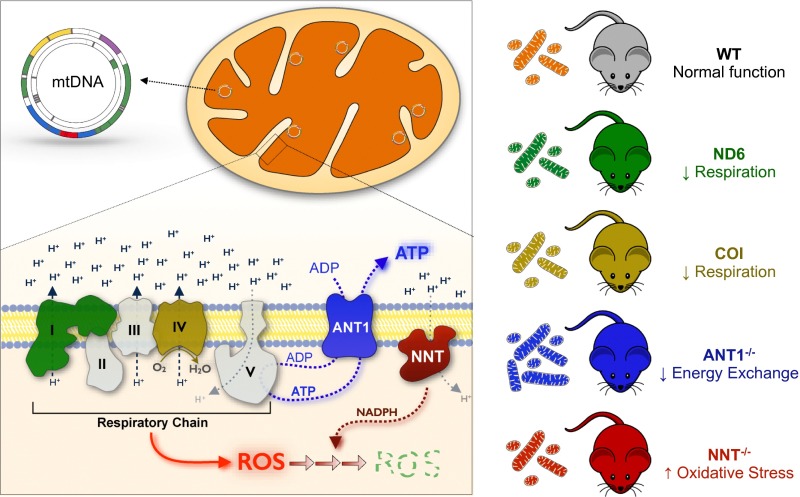
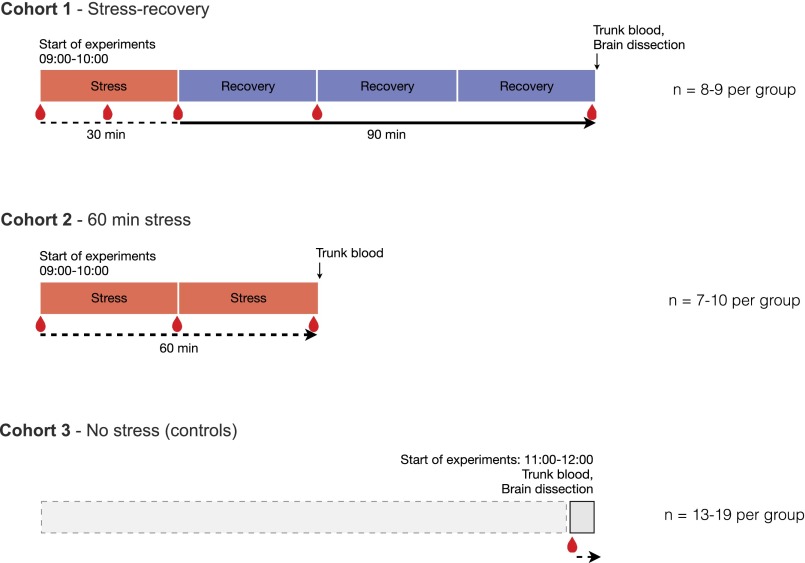
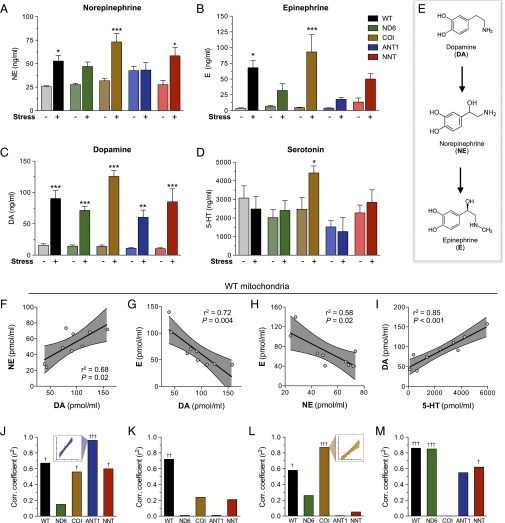
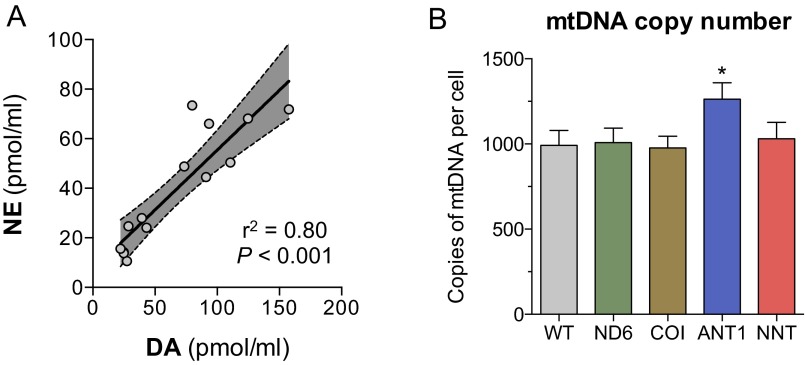
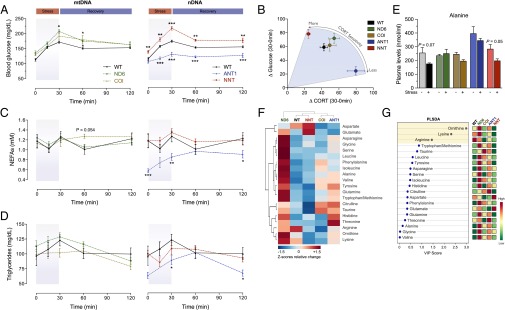
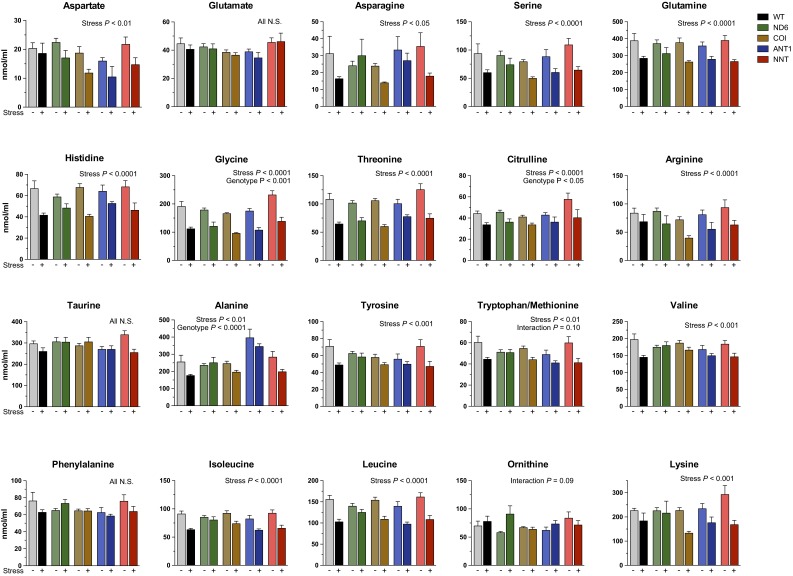
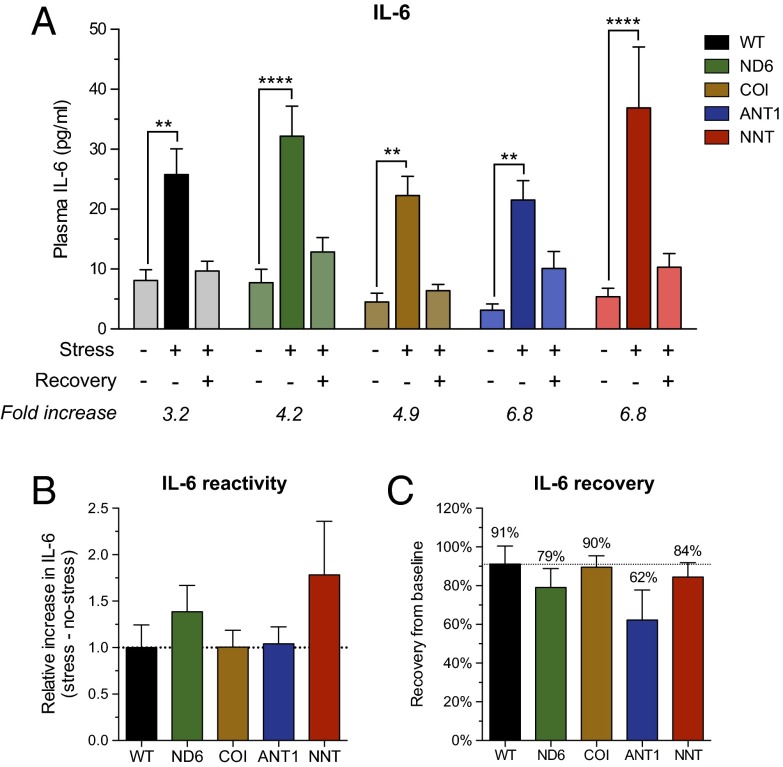
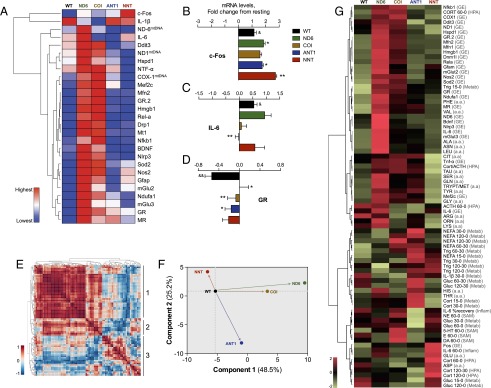
The experience of psychological stress triggers neuroendocrine, inflammatory, metabolic, and transcriptional perturbations that ultimately predispose to disease. However, the subcellular determinants of this integrated, multisystemic stress response have not been defined. Central to stress adaptation is cellular energetics, involving mitochondrial energy production and oxidative stress. We therefore hypothesized that abnormal mitochondrial functions would differentially modulate the organism's multisystemic response to psychological stress. By mutating or deleting mitochondrial genes encoded in the mtDNA [NADH dehydrogenase 6 (ND6) and cytochrome c oxidase subunit I (COI)] or nuclear DNA [adenine nucleotide translocator 1 (ANT1) and nicotinamide nucleotide transhydrogenase (NNT)], we selectively impaired mitochondrial respiratory chain function, energy exchange, and mitochondrial redox balance in mice. The resulting impact on physiological reactivity and recovery from restraint stress were then characterized. We show that mitochondrial dysfunctions altered the hypothalamic-pituitary-adrenal axis, sympathetic adrenal-medullary activation and catecholamine levels, the inflammatory cytokine IL-6, circulating metabolites, and hippocampal gene expression responses to stress. Each mitochondrial defect generated a distinct whole-body stress-response signature. These results demonstrate the role of mitochondrial energetics and redox balance as modulators of key pathophysiological perturbations previously linked to disease. This work establishes mitochondria as stress-response modulators, with implications for understanding the mechanisms of stress pathophysiology and mitochondrial diseases.
Keywords: HPA axis; catecholamines; hippocampus; mitochondria; stress reactivity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


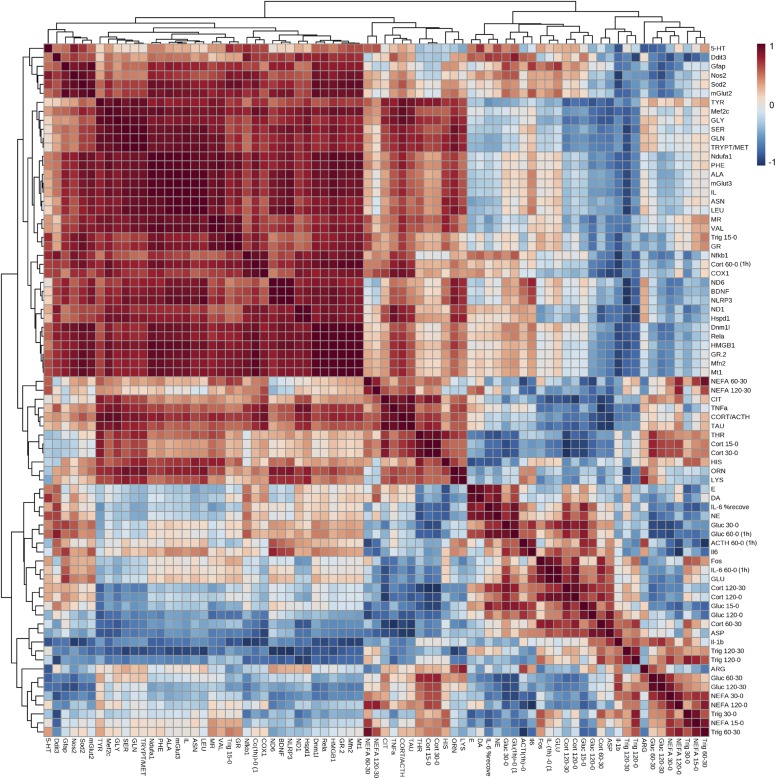

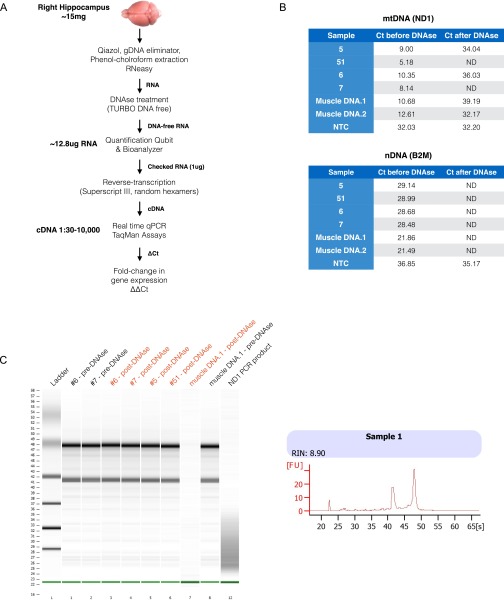

References
-
- McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44. - PubMed
-
- Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. JAMA. 2007;298(14):1685–1687. - PubMed
-
- McEwen BS. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol Rev. 2007;87(3):873–904. - PubMed
-
- Lundberg U. Stress hormones in health and illness: The roles of work and gender. Psychoneuroendocrinology. 2005;30(10):1017–1021. - PubMed
-
- Rohleder N. Stimulation of systemic low-grade inflammation by psychosocial stress. Psychosom Med. 2014;76(3):181–189. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
