

Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors
- PMID: 26628681
- PMCID: PMC4886239
- DOI: 10.1126/scisignal.aab4068
Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors
Abstract
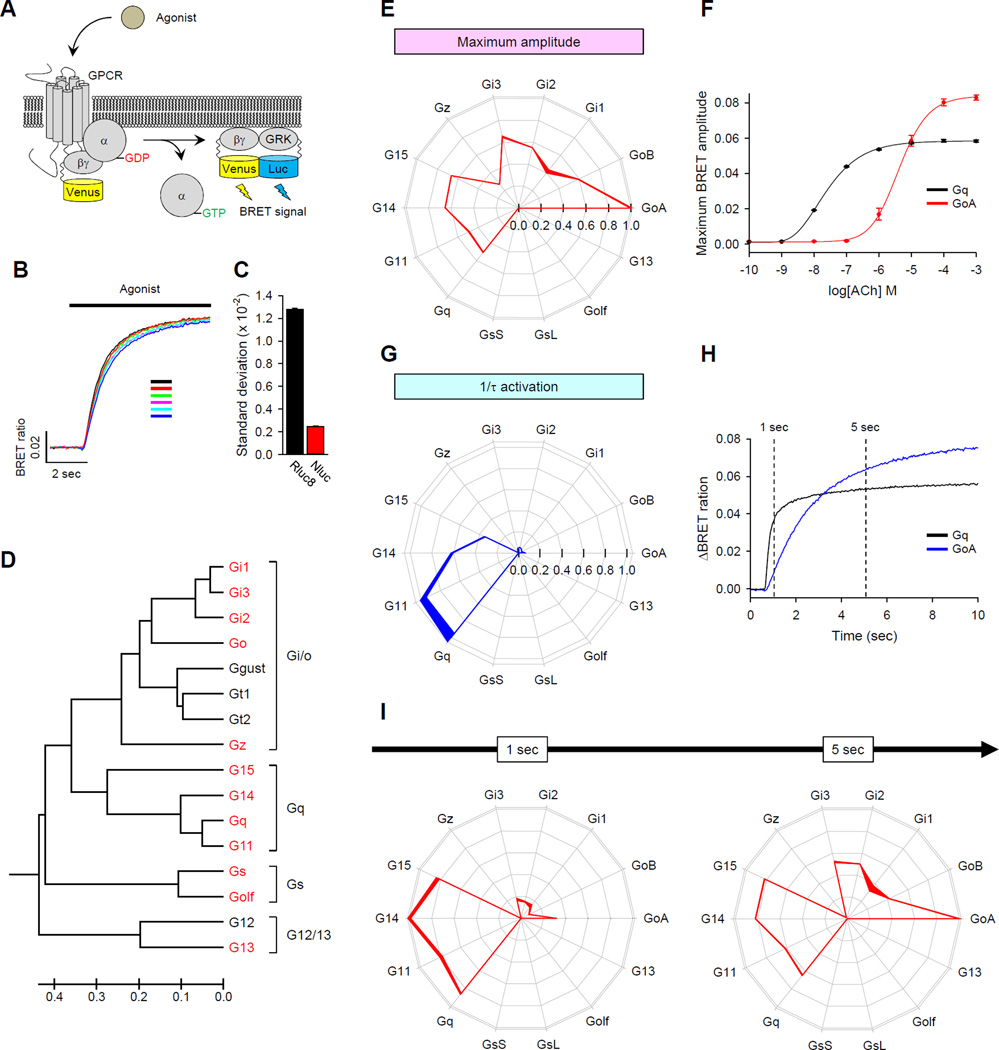
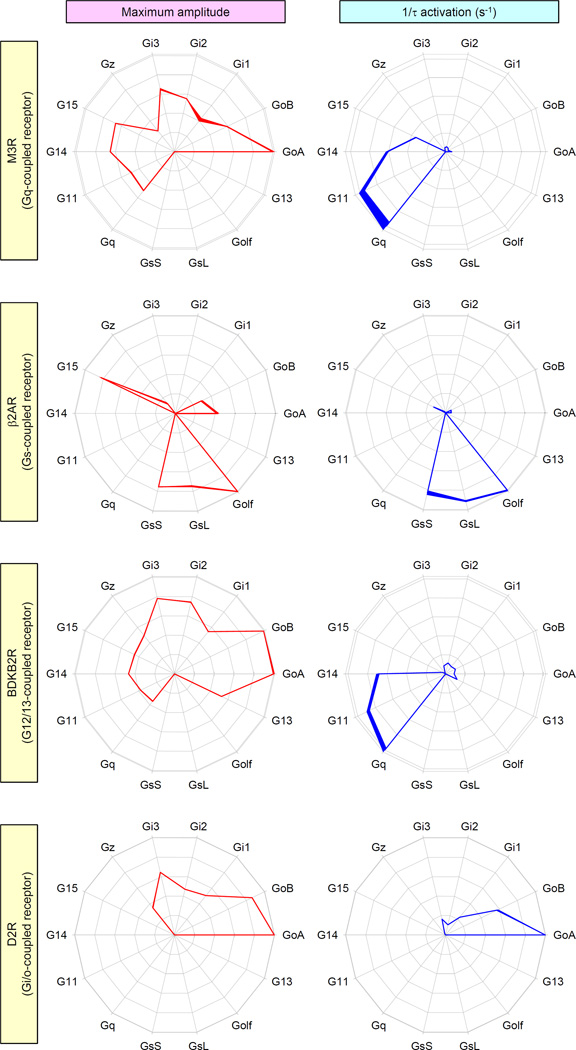
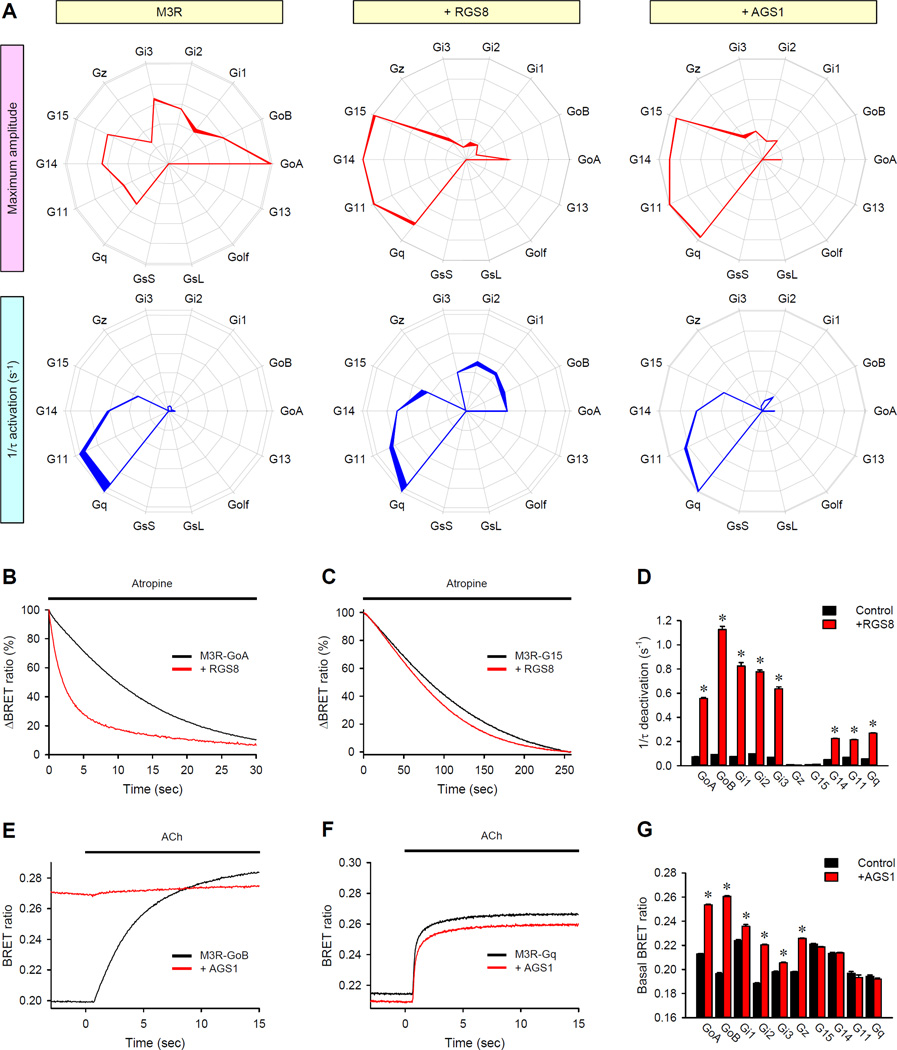
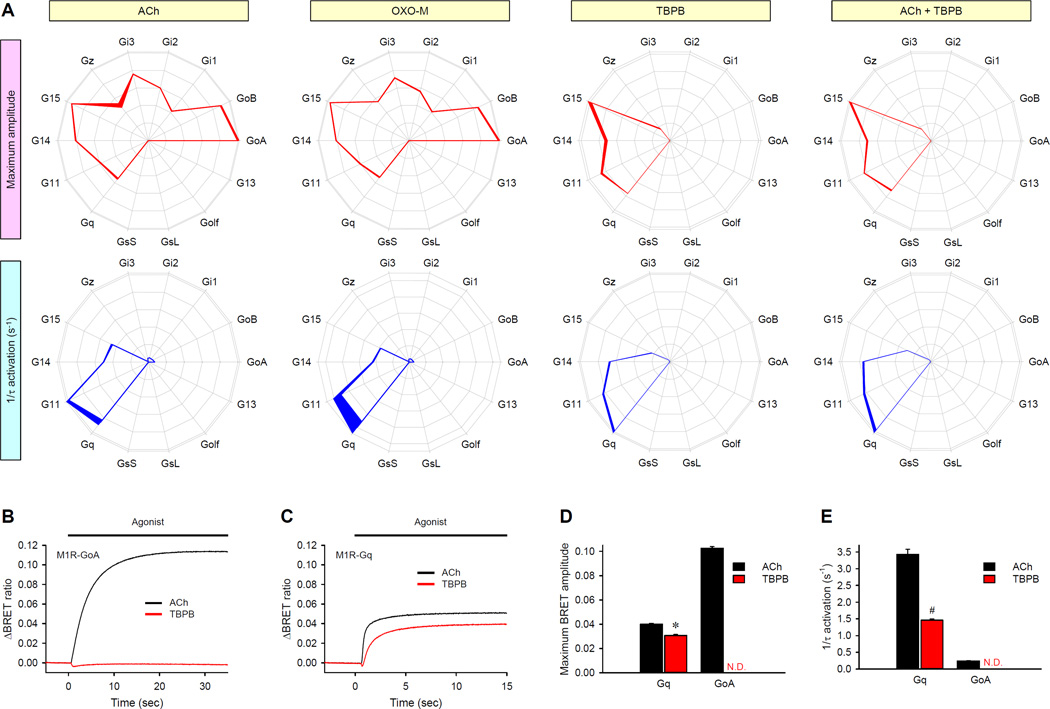
Members of the heterotrimeric guanine nucleotide-binding protein (G protein)-coupled receptor (GPCR) family play key roles in many physiological functions and are extensively exploited pharmacologically to treat diseases. Many of the diverse effects of individual GPCRs on cellular physiology are transduced by heterotrimeric G proteins, which are composed of α, β, and γ subunits. GPCRs interact with and stimulate the binding of guanosine triphosphate (GTP) to the α subunit to initiate signaling. Mammalian genomes encode 16 different G protein α subunits, each one of which has distinct properties. We developed a single-platform, optical strategy to monitor G protein activation in live cells. With this system, we profiled the coupling ability of individual GPCRs for different α subunits, simultaneously quantifying the magnitude of the signal and the rates at which the receptors activated the G proteins. We found that individual receptors engaged multiple G proteins with varying efficacy and kinetics, generating fingerprint-like profiles. Different classes of GPCR ligands, including full and partial agonists, allosteric modulators, and antagonists, distinctly affected these fingerprints to functionally bias GPCR signaling. Finally, we showed that intracellular signaling modulators further altered the G protein-coupling profiles of GPCRs, which suggests that their differential abundance may alter signaling outcomes in a cell-specific manner. These observations suggest that the diversity of the effects of GPCRs on cellular physiology may be determined by their differential engagement of multiple G proteins, coupling to which produces signals with varying signal magnitudes and activation kinetics, properties that may be exploited pharmacologically.
Copyright © 2015, American Association for the Advancement of Science.
Figures
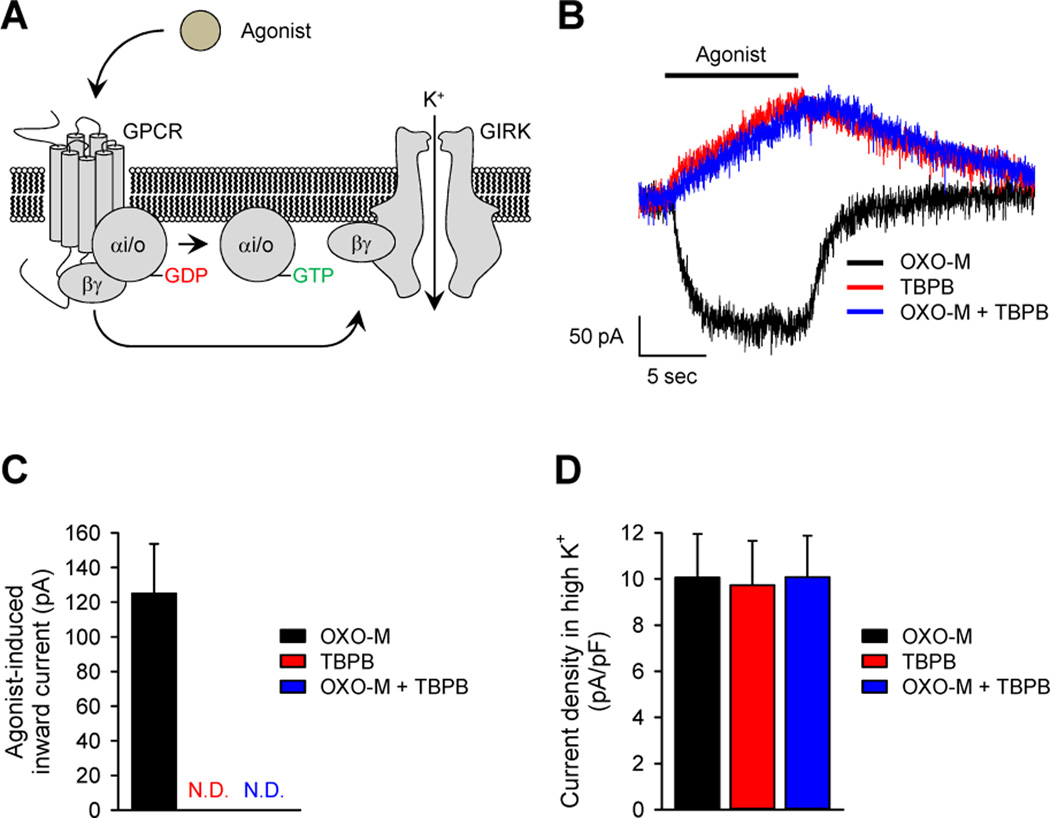
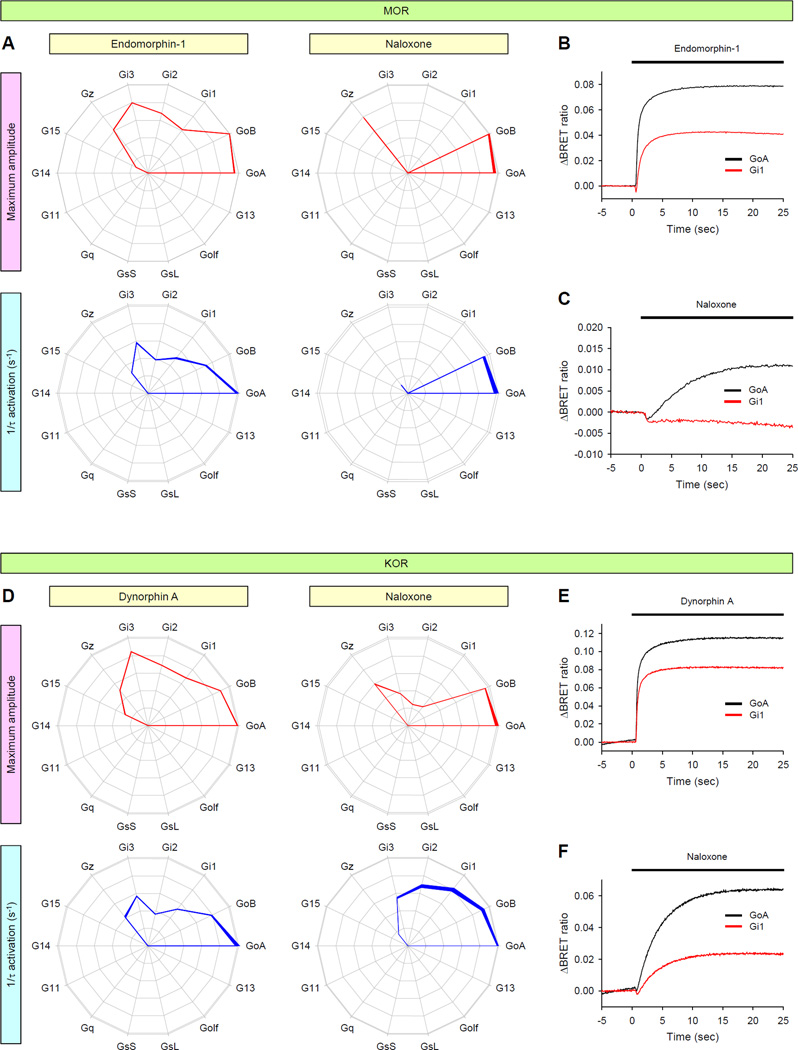
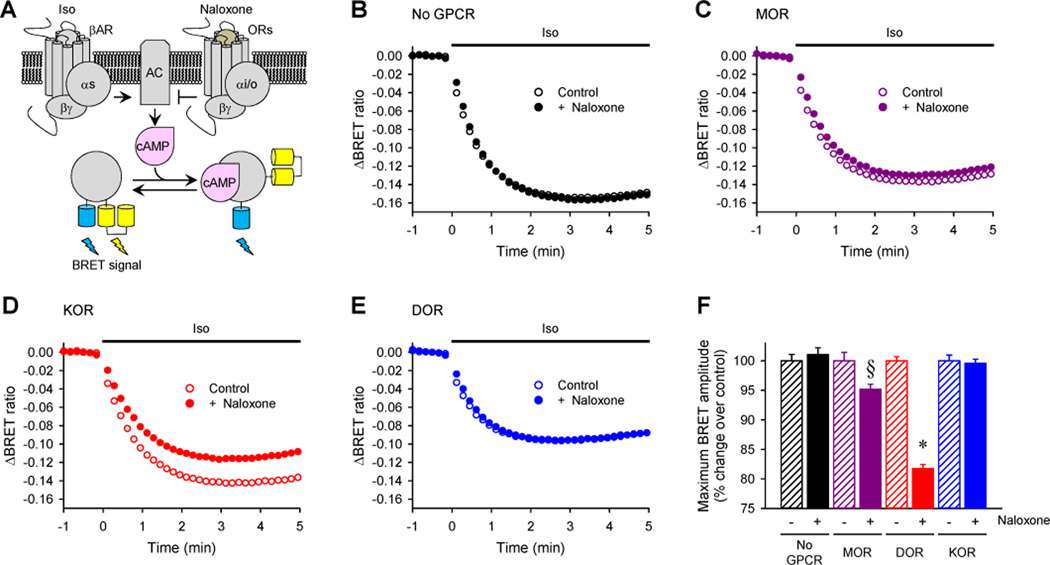
Comment in
-
Fingerprinting G protein-coupled receptor signaling.Sci Signal. 2015 Dec 1;8(405):fs20. doi: 10.1126/scisignal.aad8140. Sci Signal. 2015. PMID: 26628679
References
-
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiological reviews. 2005;85:1159–1204. - PubMed
-
- Vassart G, Costagliola S. G protein-coupled receptors: mutations and endocrine diseases. Nature reviews. Endocrinology. 2011;7:362–372. - PubMed
-
- Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat Rev Drug Discov. 2012;11:603–619. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
