

Interaction between a Domain of the Negative Regulator of the Ras-ERK Pathway, SPRED1 Protein, and the GTPase-activating Protein-related Domain of Neurofibromin Is Implicated in Legius Syndrome and Neurofibromatosis Type 1
- PMID: 26635368
- PMCID: PMC4751360
- DOI: 10.1074/jbc.M115.703710
Interaction between a Domain of the Negative Regulator of the Ras-ERK Pathway, SPRED1 Protein, and the GTPase-activating Protein-related Domain of Neurofibromin Is Implicated in Legius Syndrome and Neurofibromatosis Type 1
Abstract
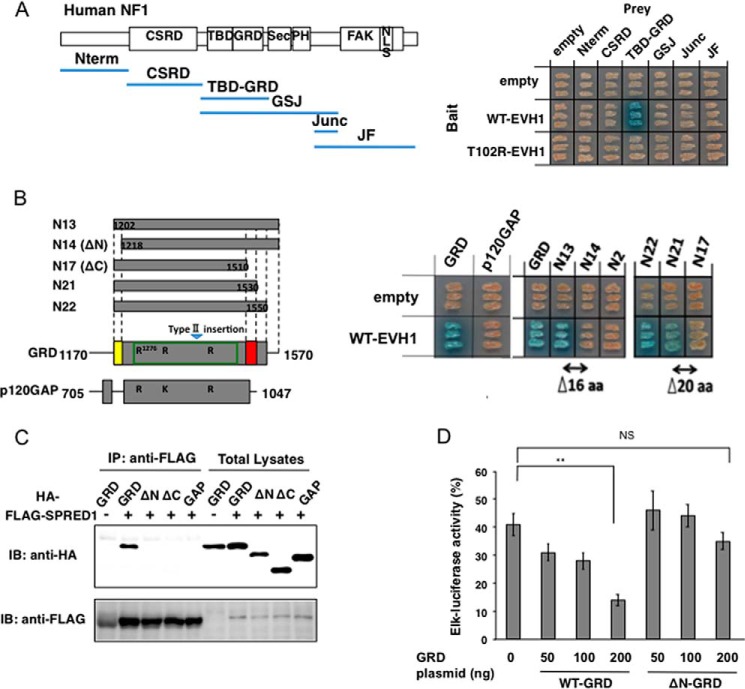
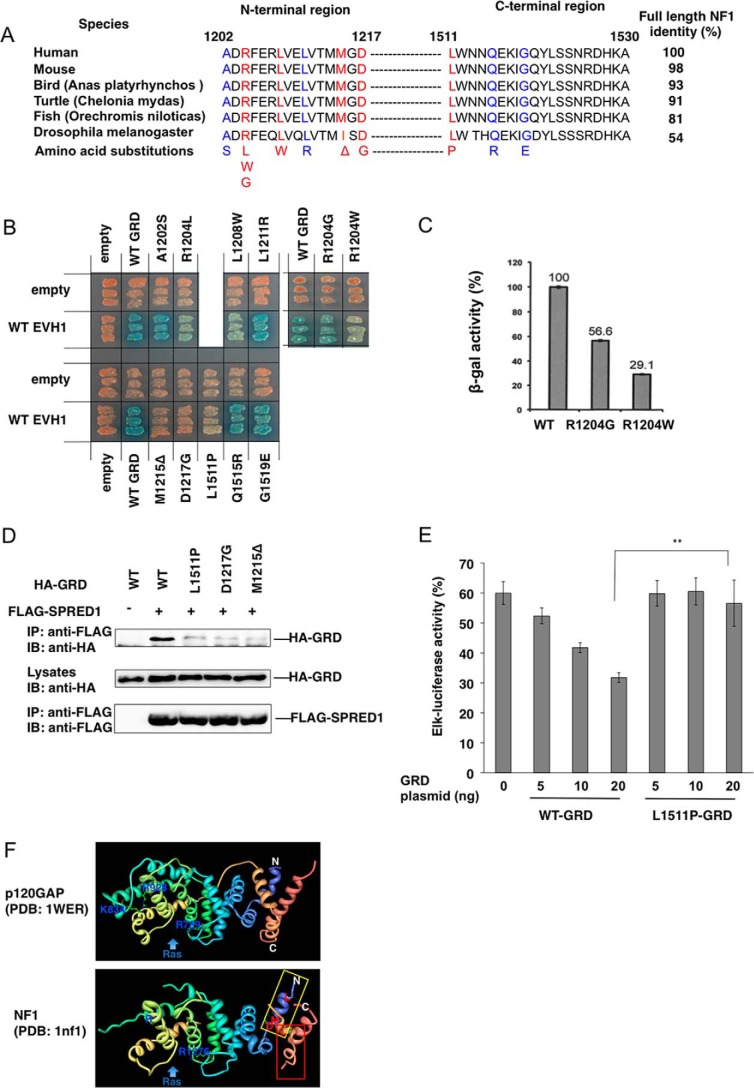
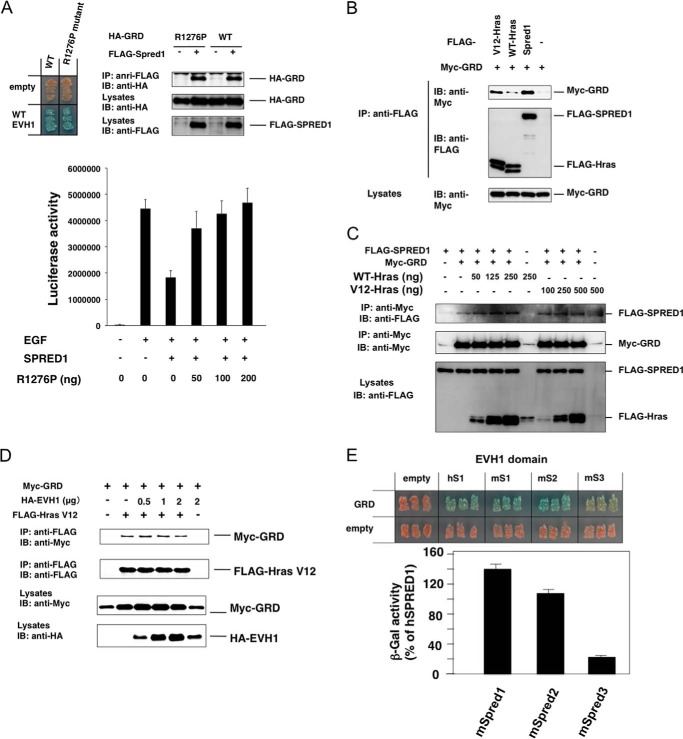
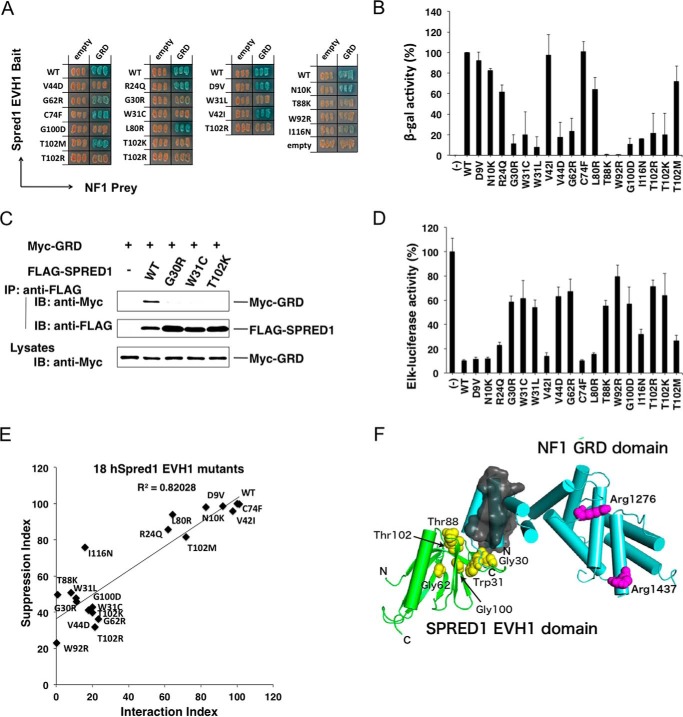
Constitutional heterozygous loss-of-function mutations in the SPRED1 gene cause a phenotype known as Legius syndrome, which consists of symptoms of multiple café-au-lait macules, axillary freckling, learning disabilities, and macrocephaly. Legius syndrome resembles a mild neurofibromatosis type 1 (NF1) phenotype. It has been demonstrated that SPRED1 functions as a negative regulator of the Ras-ERK pathway and interacts with neurofibromin, the NF1 gene product. However, the molecular details of this interaction and the effects of the mutations identified in Legius syndrome and NF1 on this interaction have not yet been investigated. In this study, using a yeast two-hybrid system and an immunoprecipitation assay in HEK293 cells, we found that the SPRED1 EVH1 domain interacts with the N-terminal 16 amino acids and the C-terminal 20 amino acids of the GTPase-activating protein (GAP)-related domain (GRD) of neurofibromin, which form two crossing α-helix coils outside the GAP domain. These regions have been shown to be dispensable for GAP activity and are not present in p120(GAP). Several mutations in these N- and C-terminal regions of the GRD in NF1 patients and pathogenic missense mutations in the EVH1 domain of SPRED1 in Legius syndrome reduced the binding affinity between the EVH1 domain and the GRD. EVH1 domain mutations with reduced binding to the GRD also disrupted the ERK suppression activity of SPRED1. These data clearly demonstrate that SPRED1 inhibits the Ras-ERK pathway by recruiting neurofibromin to Ras through the EVH1-GRD interaction, and this study also provides molecular basis for the pathogenic mutations of NF1 and Legius syndrome.
Keywords: GTPase-activating protein (GAP); Ras protein; human genetics; mitogen-activated protein kinase (MAPK); negative regulation; protein domain; signal transduction.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Yoshimura A. (2009) Regulation of cytokine signaling by the SOCS and Spred family proteins. Keio J. Med. 58, 73–83 - PubMed
-
- Bundschu K., Walter U., and Schuh K. (2007) Getting a first clue about SPRED functions. BioEssays 29, 897–907 - PubMed
-
- Kato R., Nonami A., Taketomi T., Wakioka T., Kuroiwa A., Matsuda Y., and Yoshimura A. (2003) Molecular cloning of mammalian Spred-3 which suppresses tyrosine kinase-mediated Erk activation. Biochem. Biophys. Res. Commun. 302, 767–772 - PubMed
-
- Nonami A., Taketomi T., Kimura A., Saeki K., Takaki H., Sanada T., Taniguchi K., Harada M., Kato R., and Yoshimura A. (2005) The Sprouty-related protein, Spred-1, localizes in a lipid raft/caveola and inhibits ERK activation in collaboration with caveolin-1. Genes Cells 10, 887–895 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
