

Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution
- PMID: 26638776
- PMCID: PMC4686820
- DOI: 10.1038/ncomms9866
Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution
Abstract
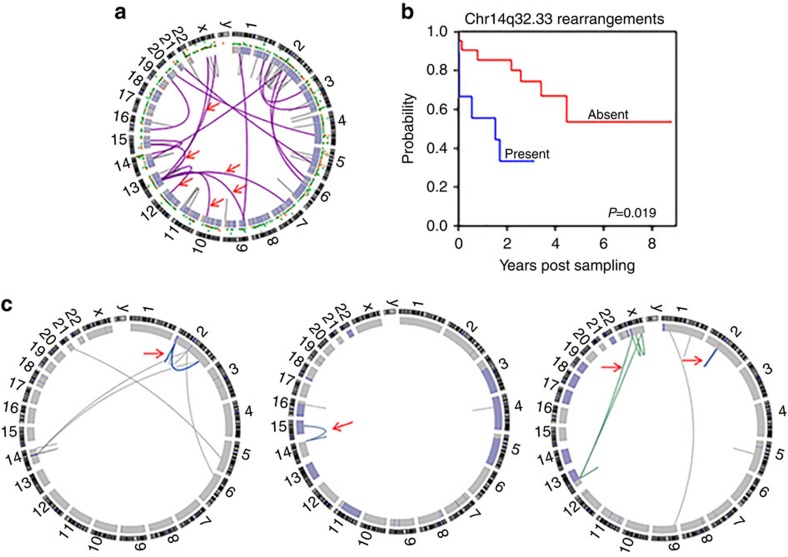
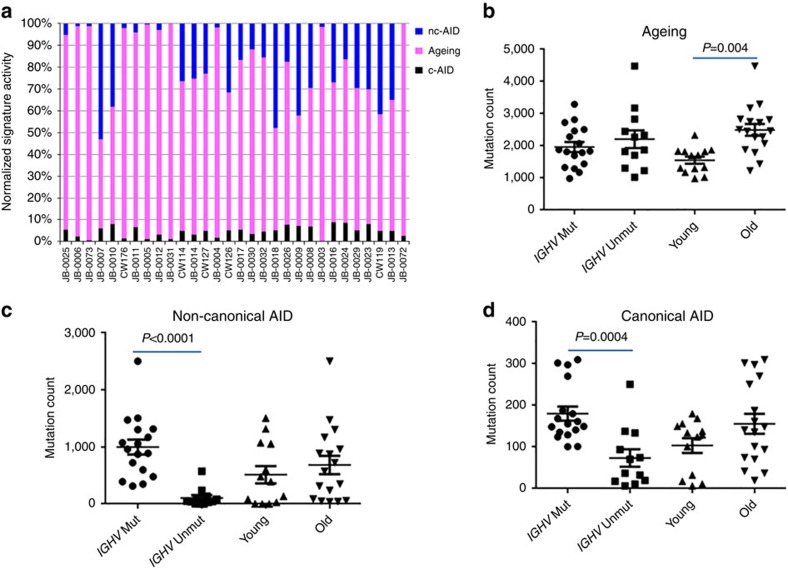
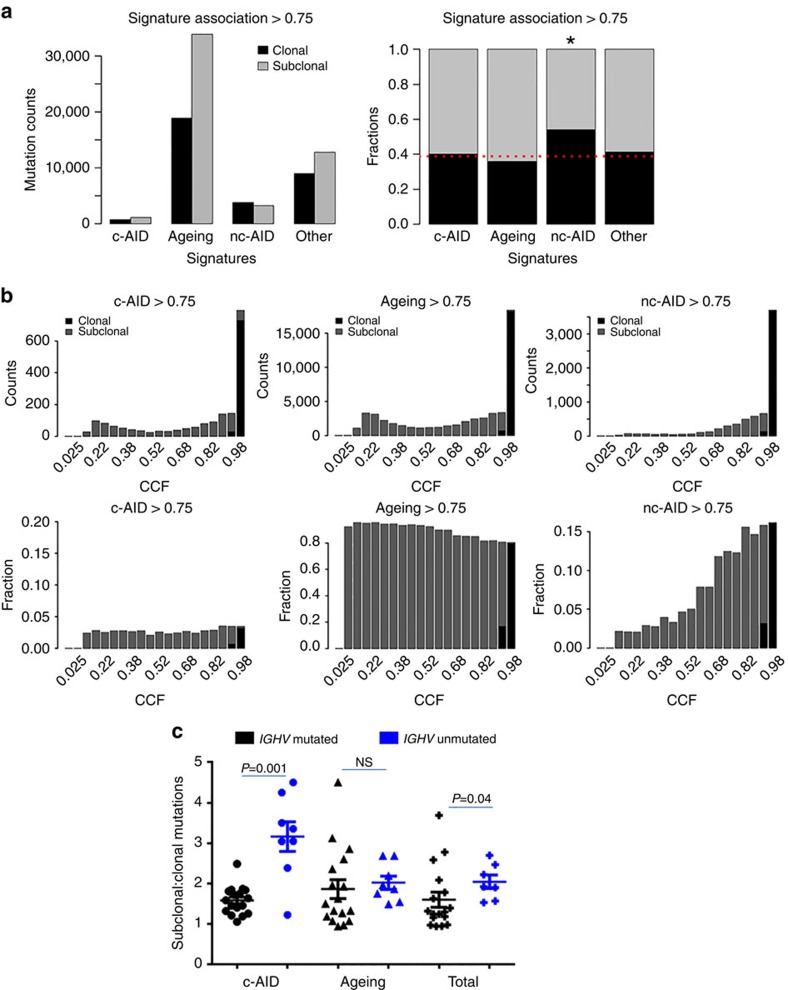
Patients with chromosome 13q deletion or normal cytogenetics represent the majority of chronic lymphocytic leukaemia (CLL) cases, yet have relatively few driver mutations. To better understand their genomic landscape, here we perform whole-genome sequencing on a cohort of patients enriched with these cytogenetic characteristics. Mutations in known CLL drivers are seen in only 33% of this cohort, and associated with normal cytogenetics and unmutated IGHV. The most commonly mutated gene in our cohort, IGLL5, shows a mutational pattern suggestive of activation-induced cytidine deaminase (AID) activity. Unsupervised analysis of mutational signatures demonstrates the activities of canonical AID (c-AID), leading to clustered mutations near active transcriptional start sites; non-canonical AID (nc-AID), leading to genome-wide non-clustered mutations, and an ageing signature responsible for most mutations. Using mutation clonality to infer time of onset, we find that while ageing and c-AID activities are ongoing, nc-AID-associated mutations likely occur earlier in tumour evolution.
Figures




References
-
- Dohner H. et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 343, 1910–1916 (2000). - PubMed
-
- Tan V. Y. & Fevotte C. Automatic relevance determination in nonnegative matrix factorization with the beta-divergence. IEEE Trans. Pattern Anal. Mach. Intell. 35, 1592–1605 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
