

Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines
- PMID: 26639779
- PMCID: PMC5458636
- DOI: 10.1038/nrmicro.2015.4
Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines
Abstract
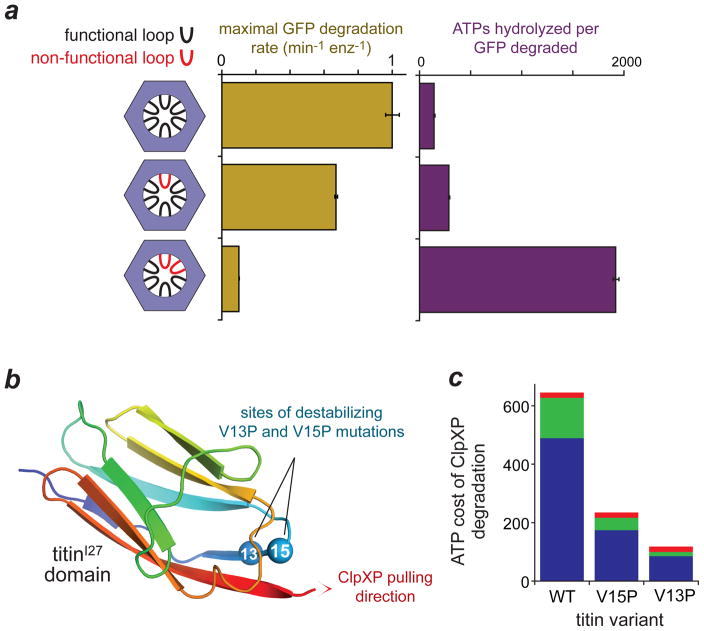
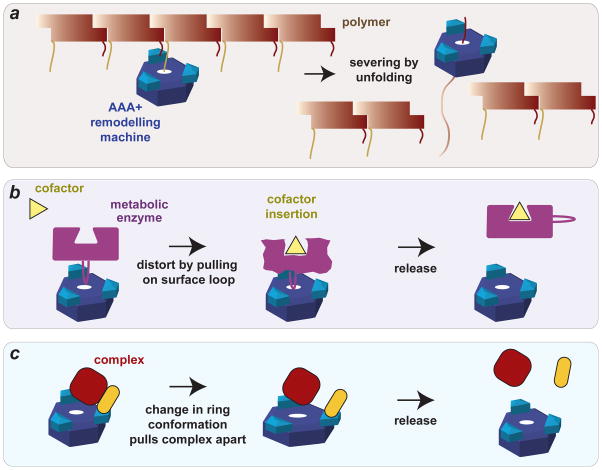
To maintain protein homeostasis, AAA+ proteolytic machines degrade damaged and unneeded proteins in bacteria, archaea and eukaryotes. This process involves the ATP-dependent unfolding of a target protein and its subsequent translocation into a self-compartmentalized proteolytic chamber. Related AAA+ enzymes also disaggregate and remodel proteins. Recent structural and biochemical studies, in combination with direct visualization of unfolding and translocation in single-molecule experiments, have illuminated the molecular mechanisms behind these processes and suggest how remodelling of macromolecular complexes by AAA+ enzymes could occur without global denaturation. In this Review, we discuss the structural and mechanistic features of AAA+ proteases and remodelling machines, focusing on the bacterial ClpXP and ClpX as paradigms. We also consider the potential of these enzymes as antibacterial targets and outline future challenges for the field.
Conflict of interest statement
The authors declare no competing interests.
Figures
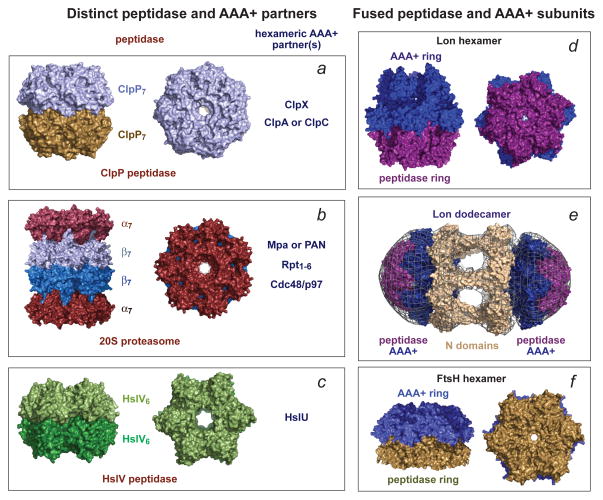
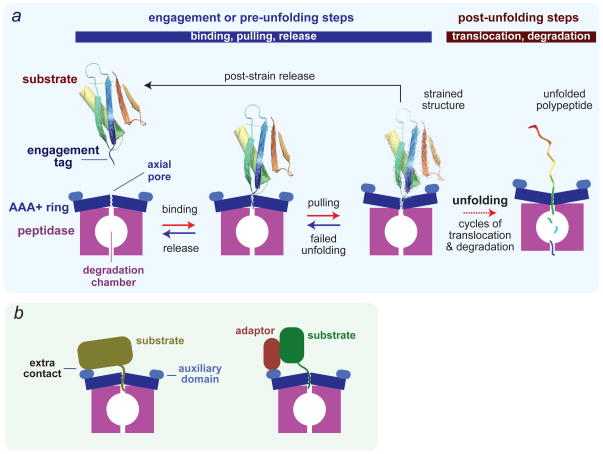
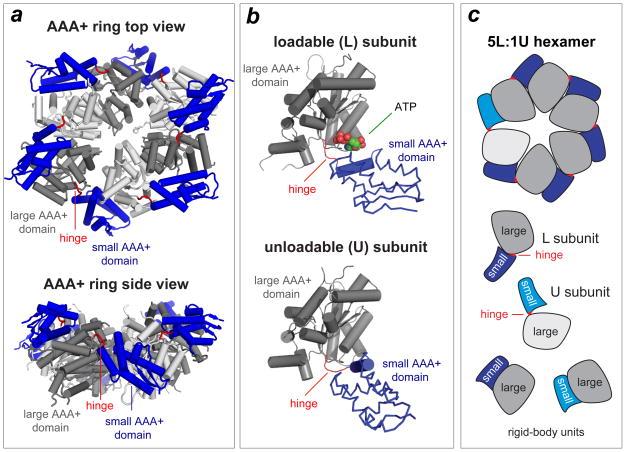
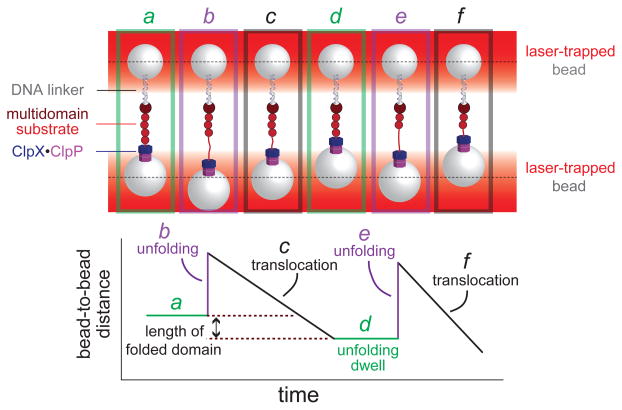
References
-
- Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem. 2014;83:553–584. - PubMed
-
- Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. - PubMed
-
- Striebel F, Kress W, Weber-Ban E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol. 2009;19:209–217. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
