

TGF-β Controls miR-181/ERK Regulatory Network during Retinal Axon Specification and Growth
- PMID: 26641497
- PMCID: PMC4671616
- DOI: 10.1371/journal.pone.0144129
TGF-β Controls miR-181/ERK Regulatory Network during Retinal Axon Specification and Growth
Abstract
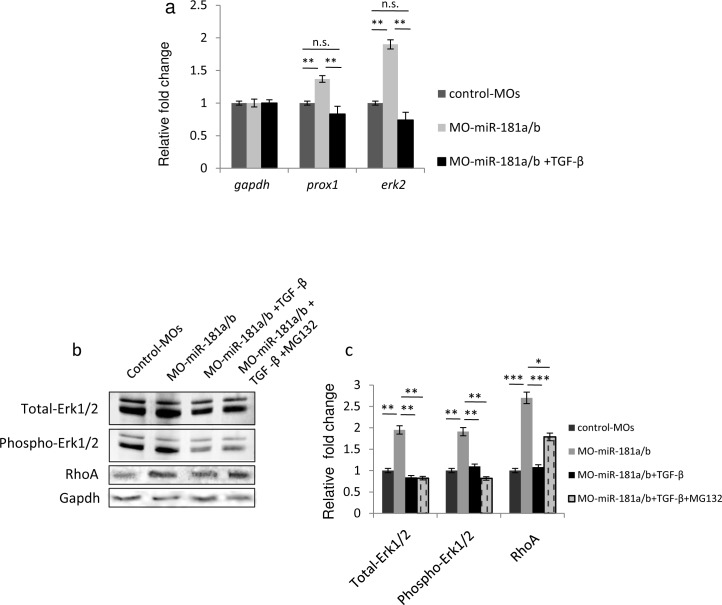
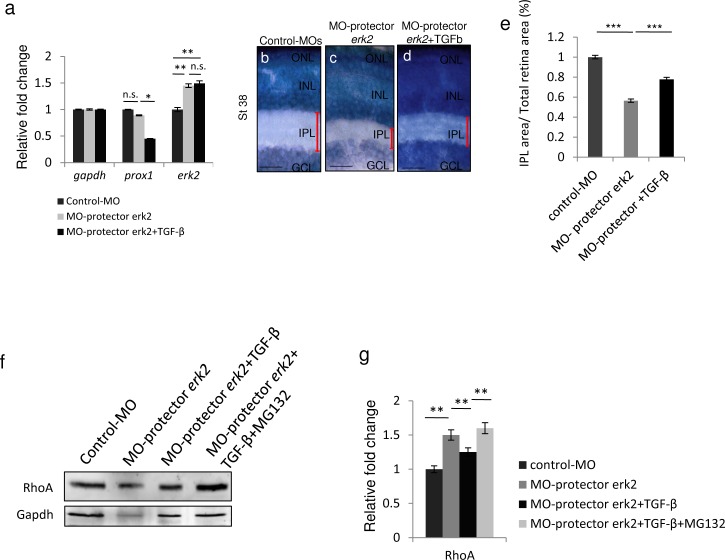
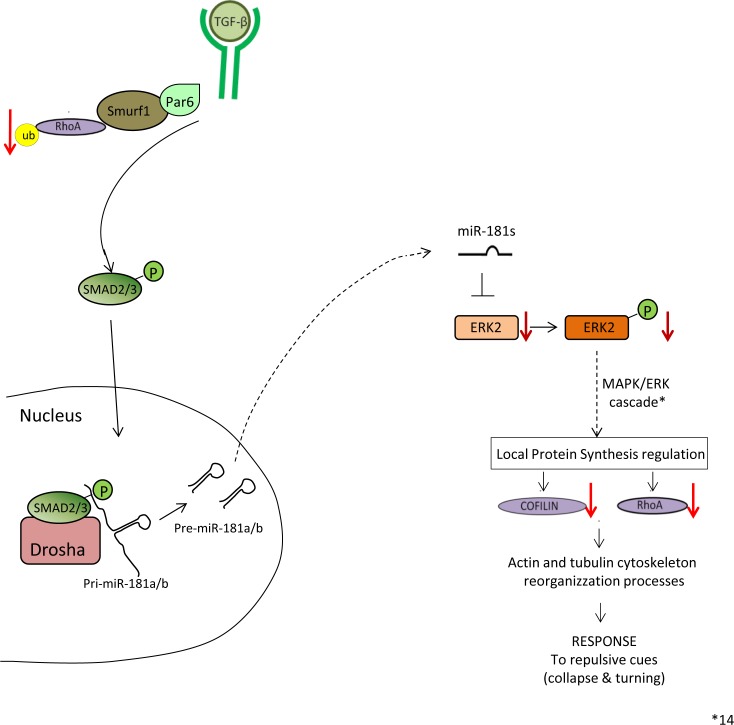
Retinal axon specification and growth are critically sensitive to the dosage of numerous signaling molecules and transcription factors. Subtle variations in the expression levels of key molecules may result in a variety of axonal growth anomalies. miR-181a and miR-181b are two eye-enriched microRNAs whose inactivation in medaka fish leads to alterations of the proper establishment of connectivity and function in the visual system. miR-181a/b are fundamental regulators of MAPK signaling and their role in retinal axon growth and specification is just beginning to be elucidated. Here we demonstrate that miR-181a/b are key nodes in the interplay between TGF-β and MAPK/ERK within the functional pathways that control retinal axon specification and growth. Using a variety of in vivo and in vitro approaches in medaka fish, we demonstrate that TGF-β signaling controls the miR-181/ERK regulatory network, which in turn strengthens the TGF-β-mediated regulation of RhoA degradation. Significantly, these data uncover the role of TGF-β signaling in vivo, for the first time, in defining the correct wiring and assembly of functional retina neural circuits and further highlight miR-181a/b as key factors in axon specification and growth.
Conflict of interest statement
Figures
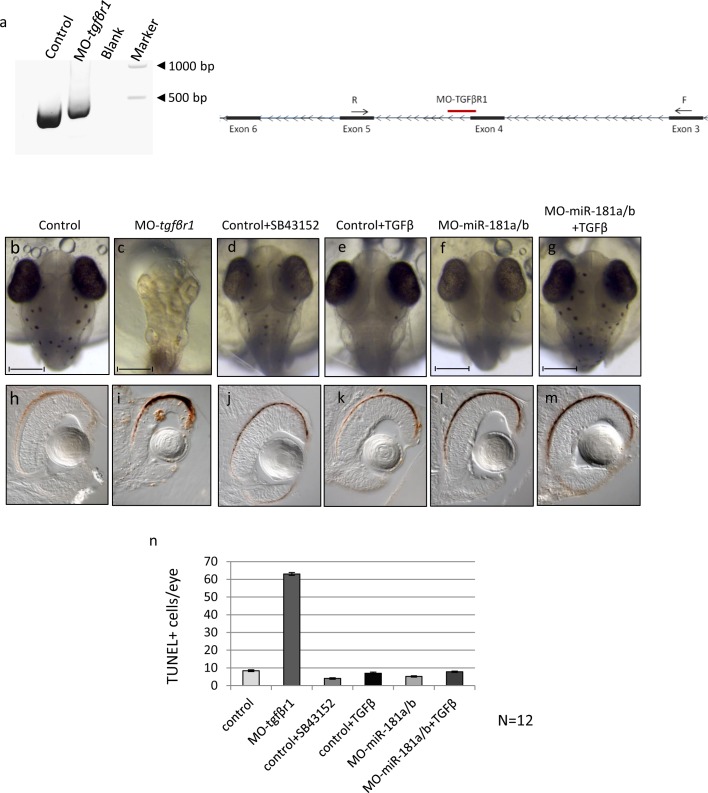
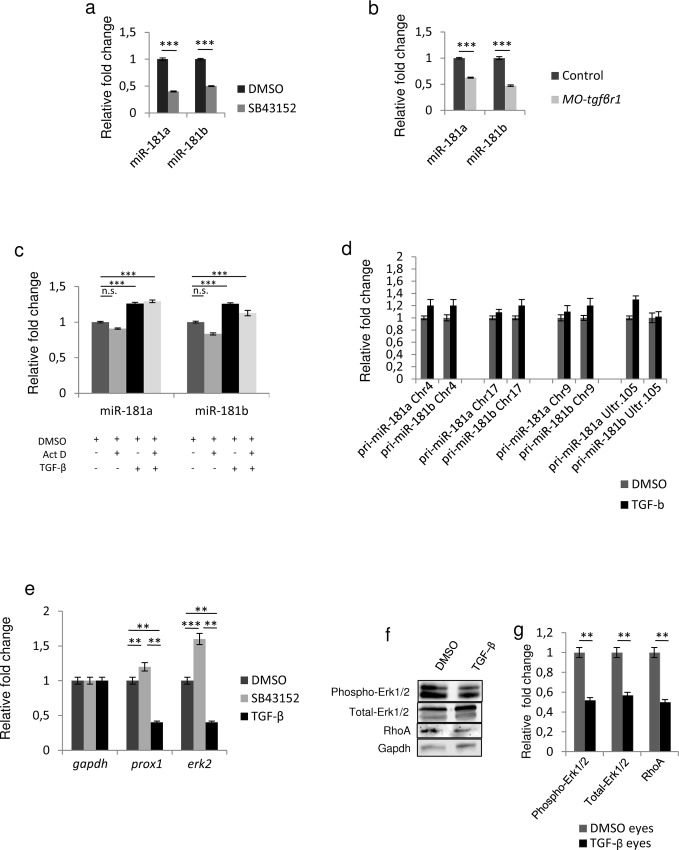
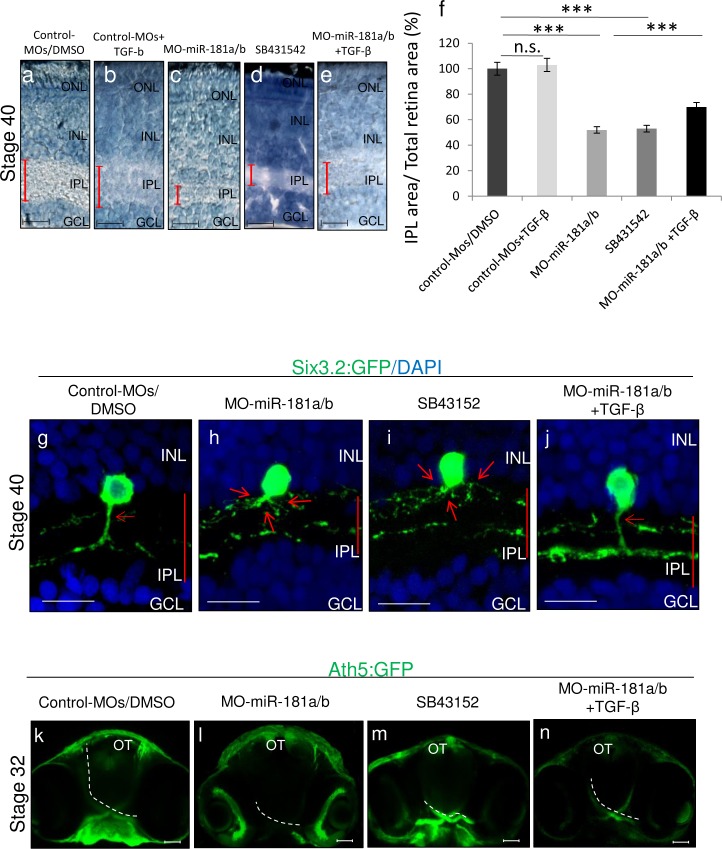
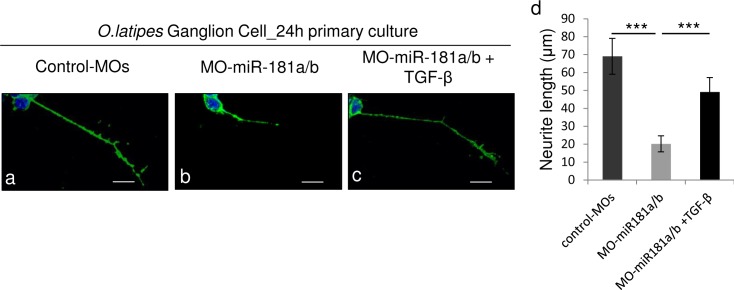
References
-
- Murali D, Kawaguchi-Niida M, Deng CX, Furuta Y. Smad4 is required predominantly in the developmental processes dependent on the BMP branch of the TGF-beta signaling system in the embryonic mouse retina. Investigative ophthalmology & visual science. 2011;52(6):2930–7. 10.1167/iovs.10-5940 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
