

BRD4 is a novel therapeutic target for liver fibrosis
- PMID: 26644586
- PMCID: PMC4697417
- DOI: 10.1073/pnas.1522163112
BRD4 is a novel therapeutic target for liver fibrosis
Abstract
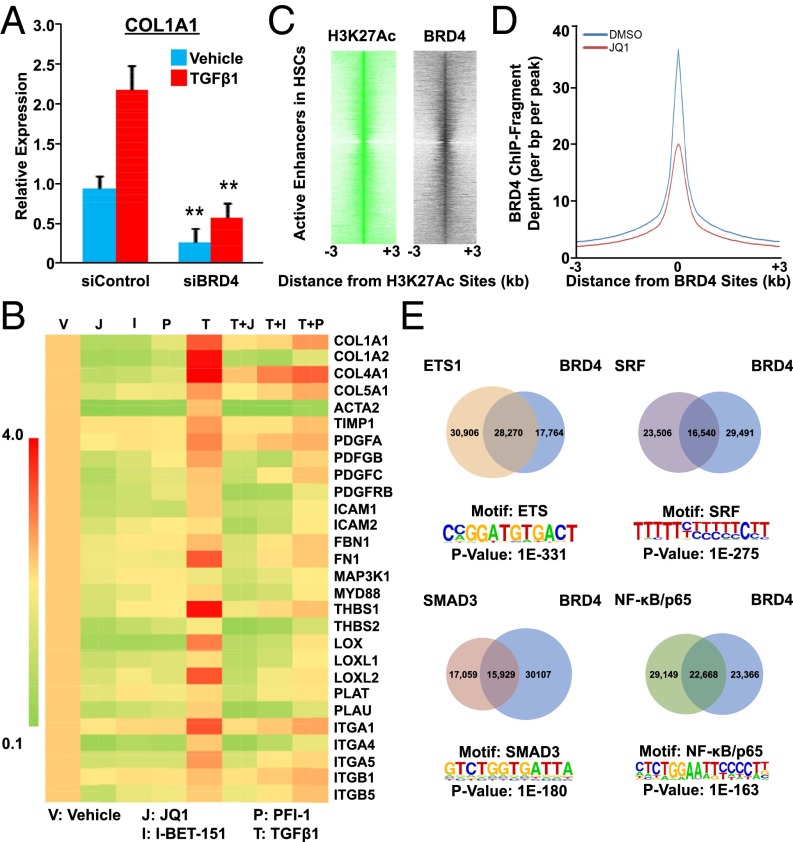
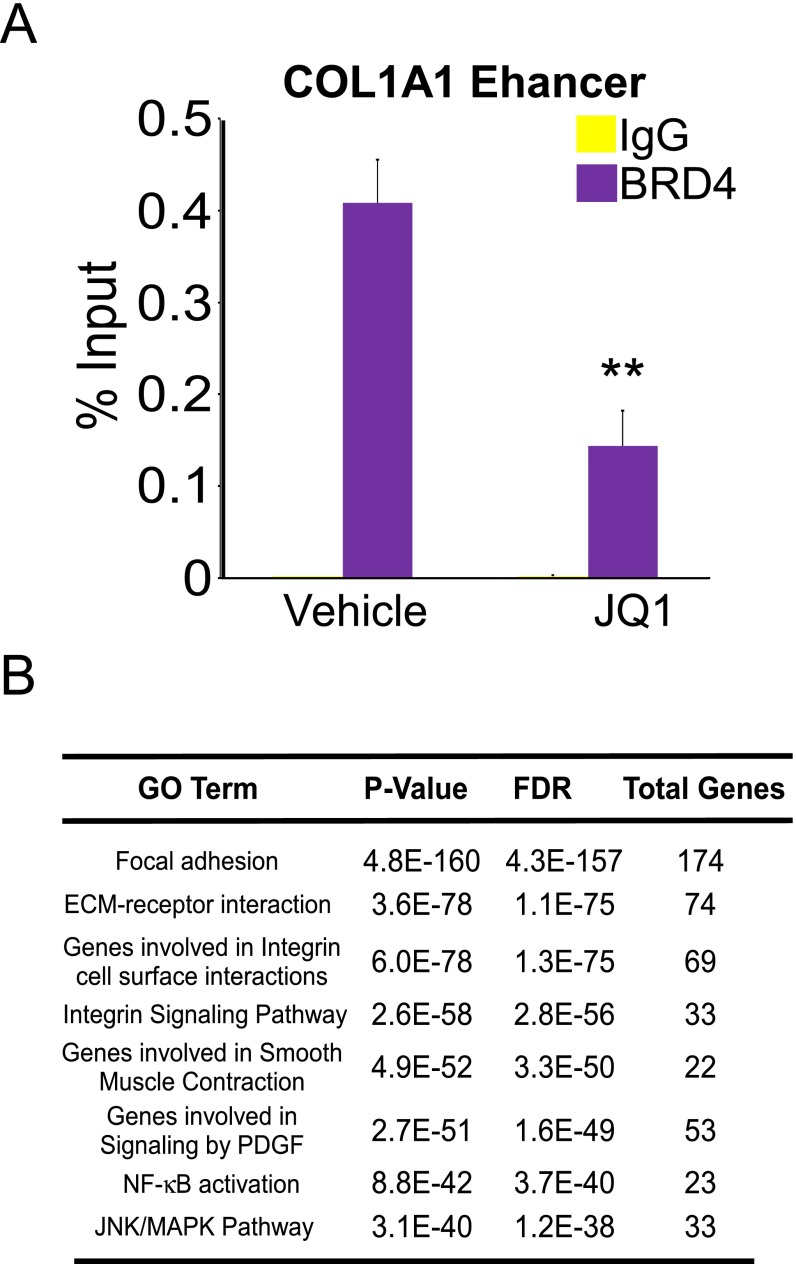
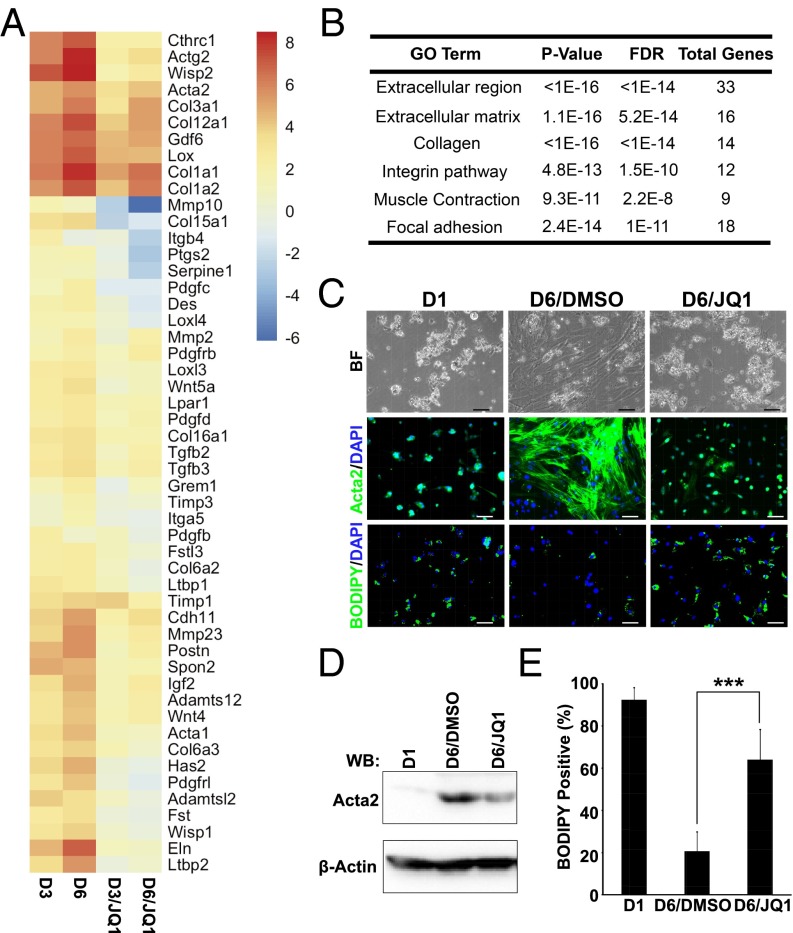
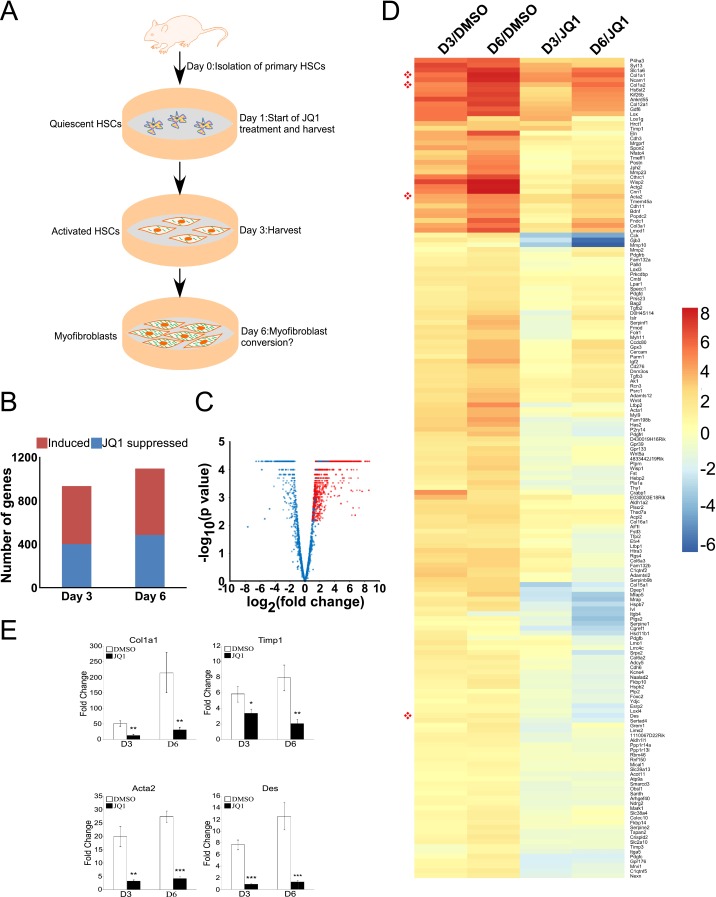
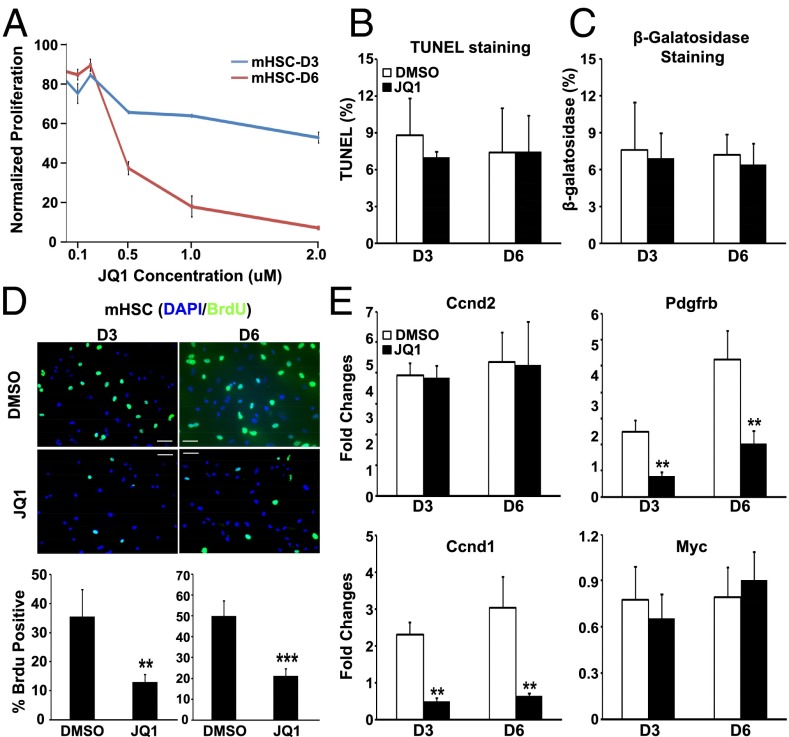
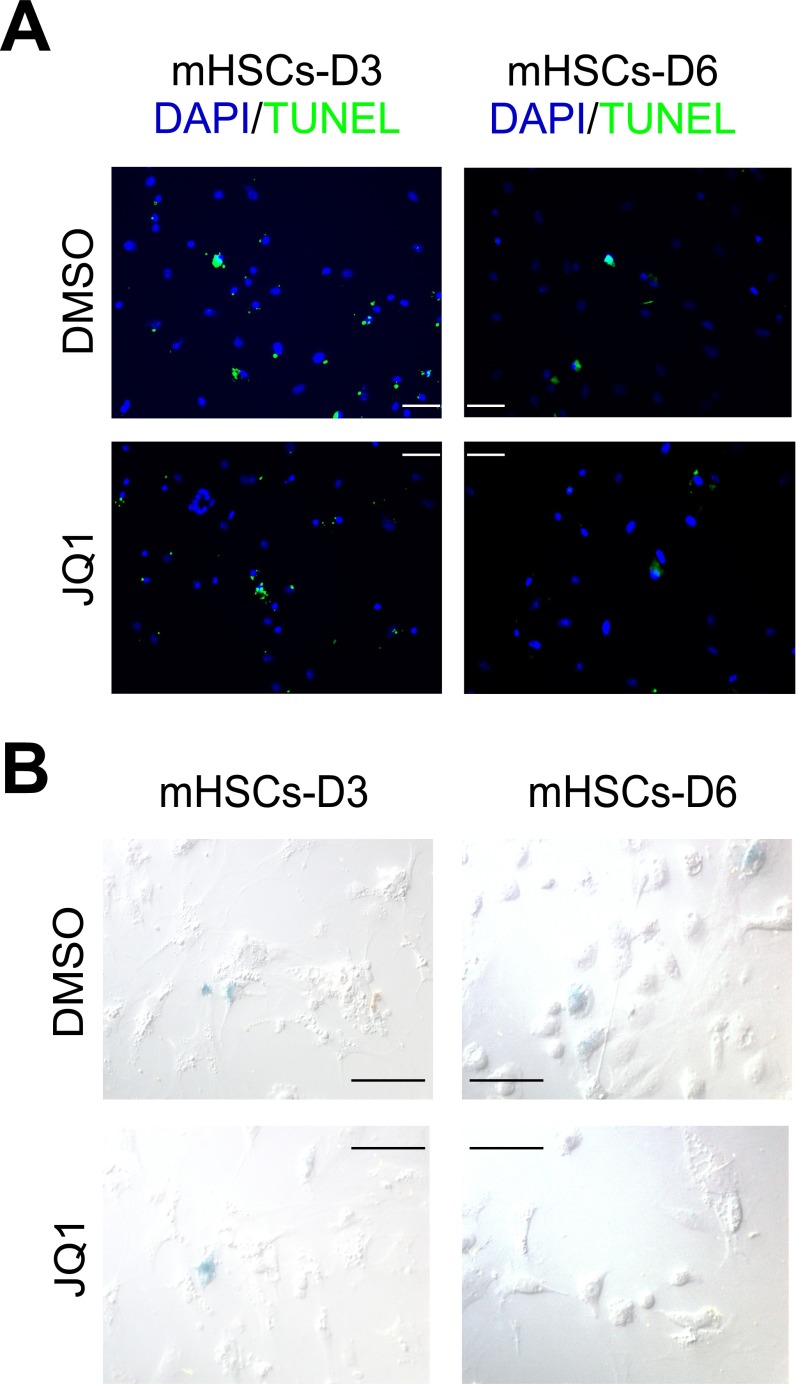
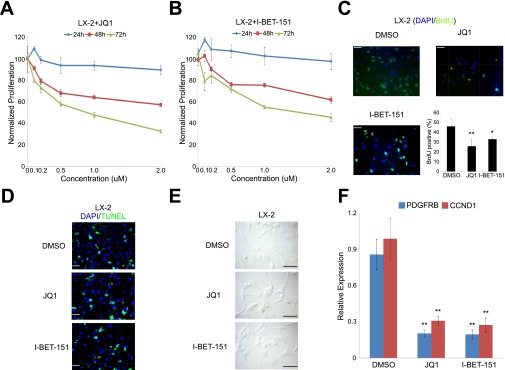
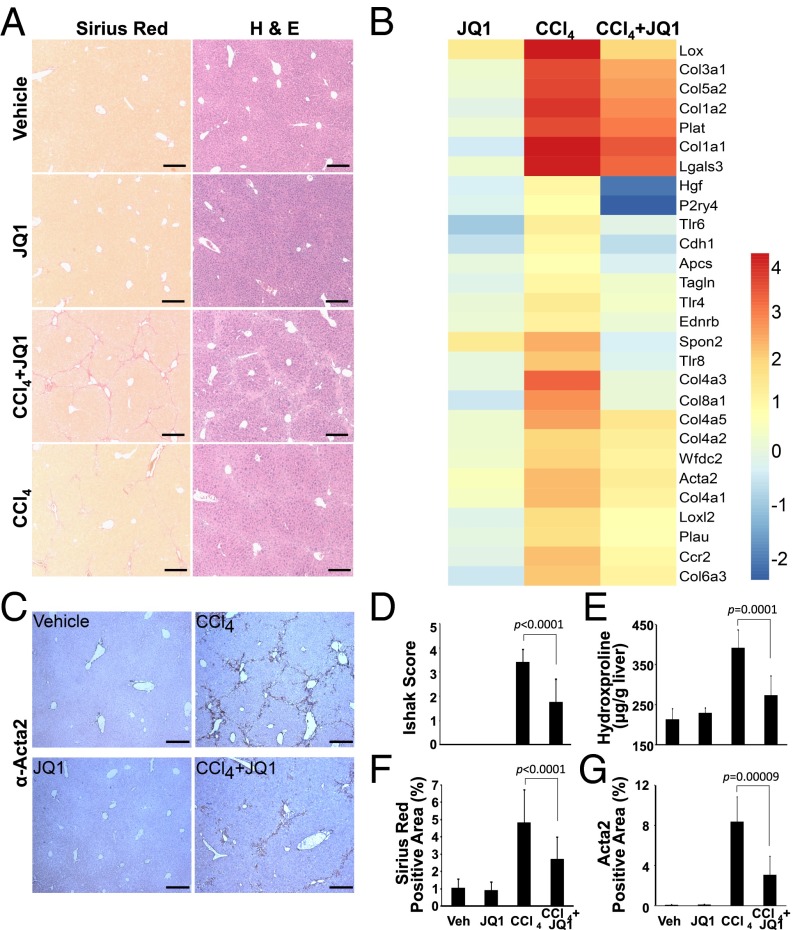
Liver fibrosis is characterized by the persistent deposition of extracellular matrix components by hepatic stellate cell (HSC)-derived myofibroblasts. It is the histological manifestation of progressive, but reversible wound-healing processes. An unabated fibrotic response results in chronic liver disease and cirrhosis, a pathological precursor of hepatocellular carcinoma. We report here that JQ1, a small molecule inhibitor of bromodomain-containing protein 4 (BRD4), a member of bromodomain and extraterminal (BET) proteins, abrogate cytokine-induced activation of HSCs. Cistromic analyses reveal that BRD4 is highly enriched at enhancers associated with genes involved in multiple profibrotic pathways, where BRD4 is colocalized with profibrotic transcription factors. Furthermore, we show that JQ1 is not only protective, but can reverse the fibrotic response in carbon tetrachloride-induced fibrosis in mouse models. Our results implicate that BRD4 can act as a global genomic regulator to direct the fibrotic response through its coordinated regulation of myofibroblast transcription. This suggests BRD4 as a potential therapeutic target for patients with fibrotic complications.
Keywords: BET inhibitor; BRD4; antifibrotic therapy; hepatic stellate cell; liver fibrosis.
Conflict of interest statement
Conflict of interest statement: N.D., R.T.Y., M.H.S., C.L., M.D., and R.M.E. are coinventors of technologies related to methods of treating fibrosis using BET inhibitors and may be entitled to royalties.
Figures
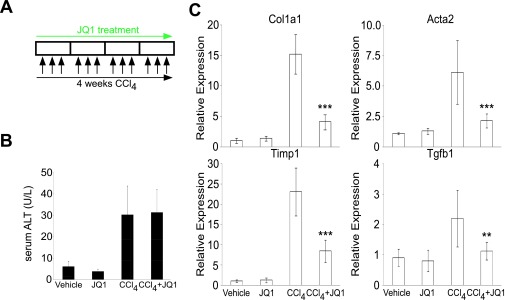
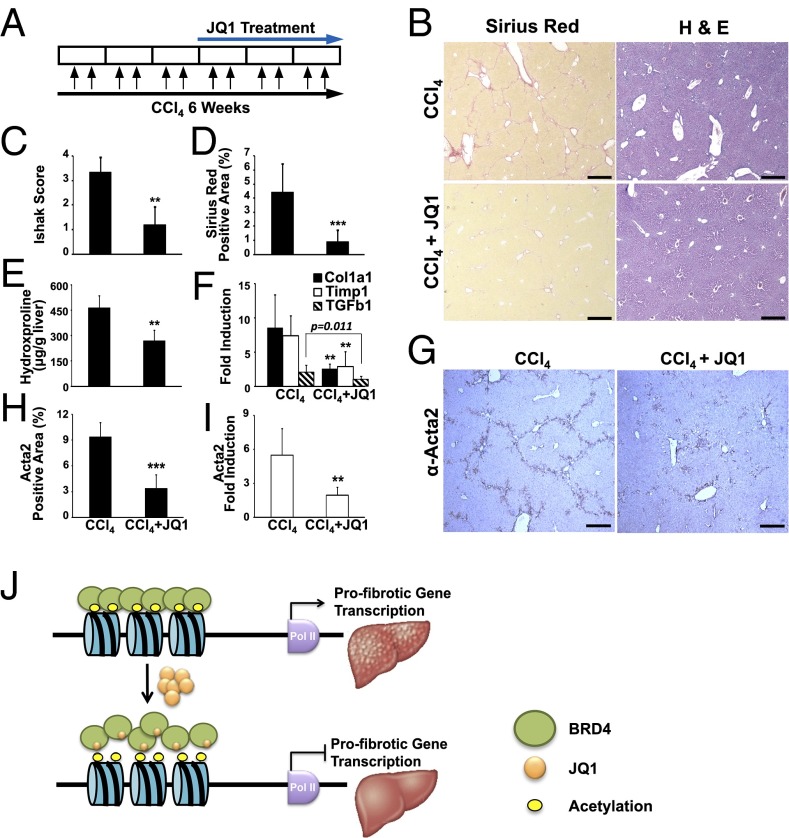
Comment in
-
Liver: Regulating profibrotic transcription.Nat Rev Gastroenterol Hepatol. 2016 Feb;13(2):62-3. doi: 10.1038/nrgastro.2015.222. Epub 2015 Dec 23. Nat Rev Gastroenterol Hepatol. 2016. PMID: 26695083 No abstract available.
References
-
- Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: Nearing the starting line. Sci Transl Med. 2013;5(167):167sr1. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- SRA/SRP043435
Grants and funding
- P01 HL088093/HL/NHLBI NIH HHS/United States
- R01 HL105278/HL/NHLBI NIH HHS/United States
- CA014195/CA/NCI NIH HHS/United States
- HL088093/HL/NHLBI NIH HHS/United States
- K01DK102867/DK/NIDDK NIH HHS/United States
- P42 ES010337/ES/NIEHS NIH HHS/United States
- T32 CA009370/CA/NCI NIH HHS/United States
- DK090962/DK/NIDDK NIH HHS/United States
- R37 DK057978/DK/NIDDK NIH HHS/United States
- HL105278/HL/NHLBI NIH HHS/United States
- K01 DK102867/DK/NIDDK NIH HHS/United States
- DK057978/DK/NIDDK NIH HHS/United States
- R24 DK090962/DK/NIDDK NIH HHS/United States
- ES010337/ES/NIEHS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 DK057978/DK/NIDDK NIH HHS/United States
- P30 CA014195/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
