

Optimizing cancer genome sequencing and analysis
- PMID: 26645048
- PMCID: PMC4669575
- DOI: 10.1016/j.cels.2015.08.015
Optimizing cancer genome sequencing and analysis
Abstract
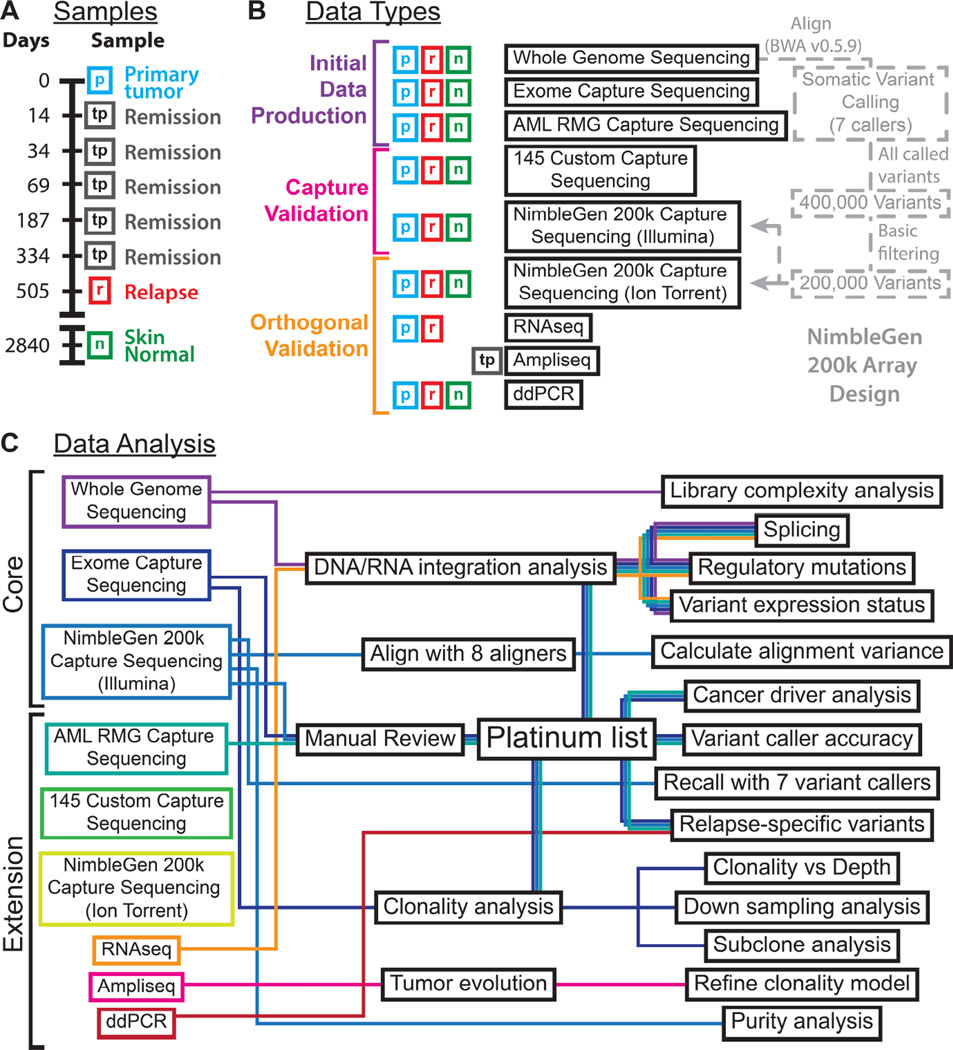
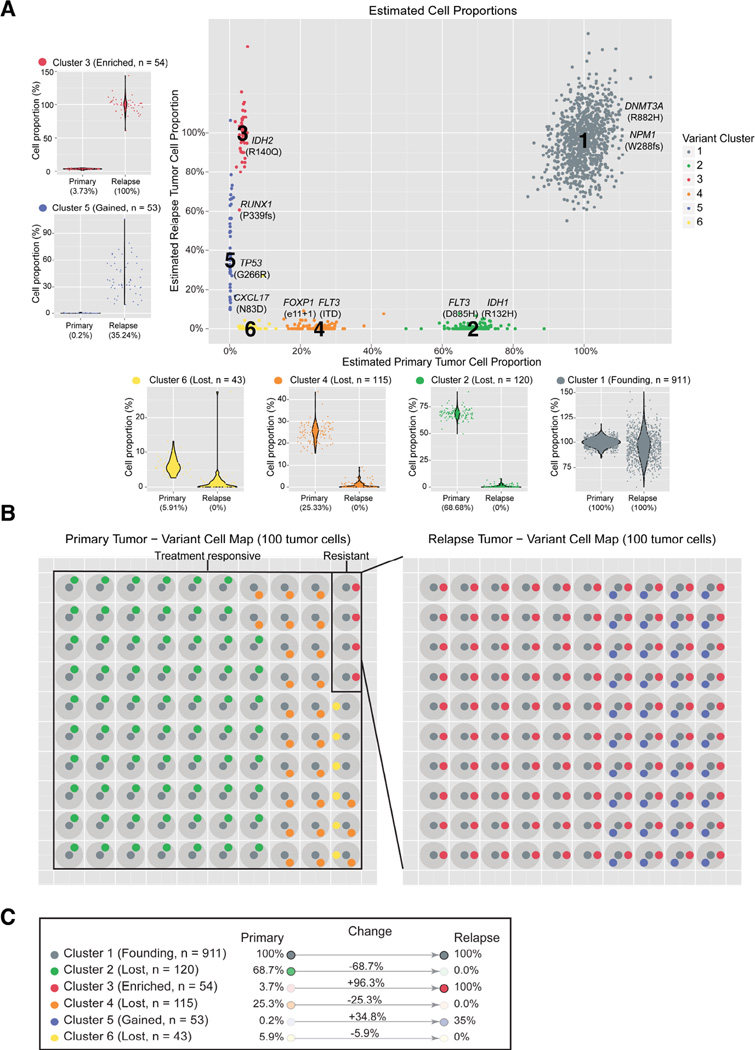
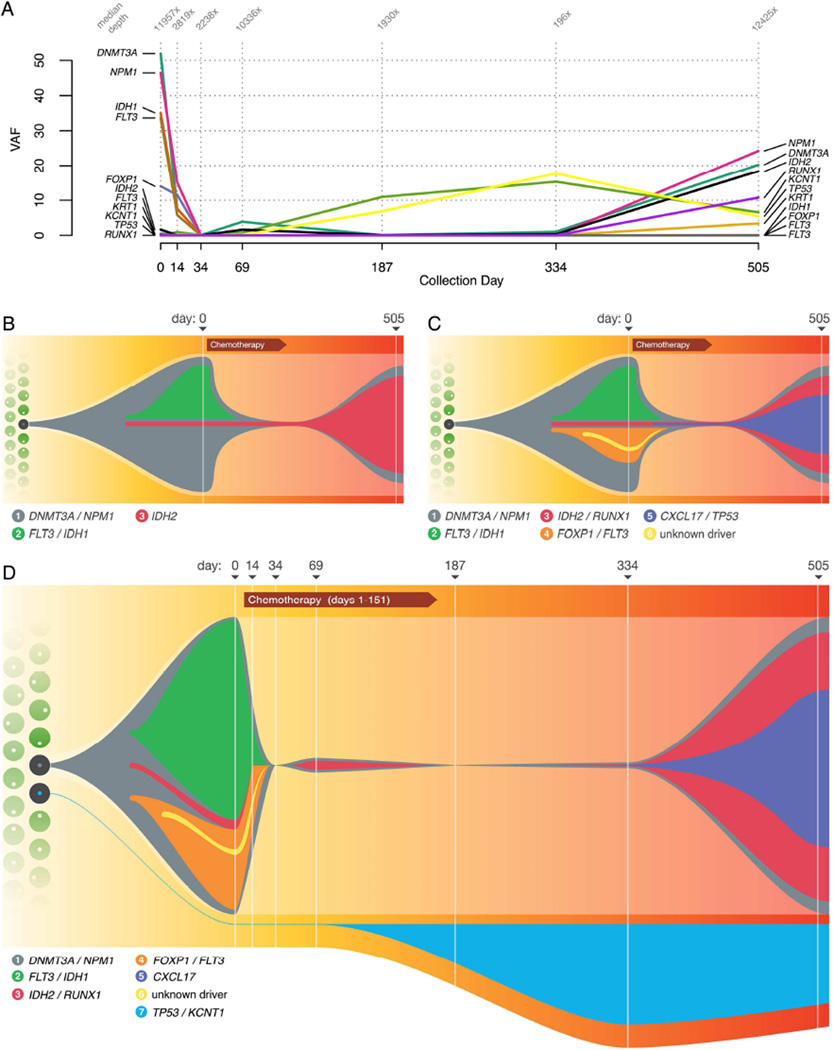
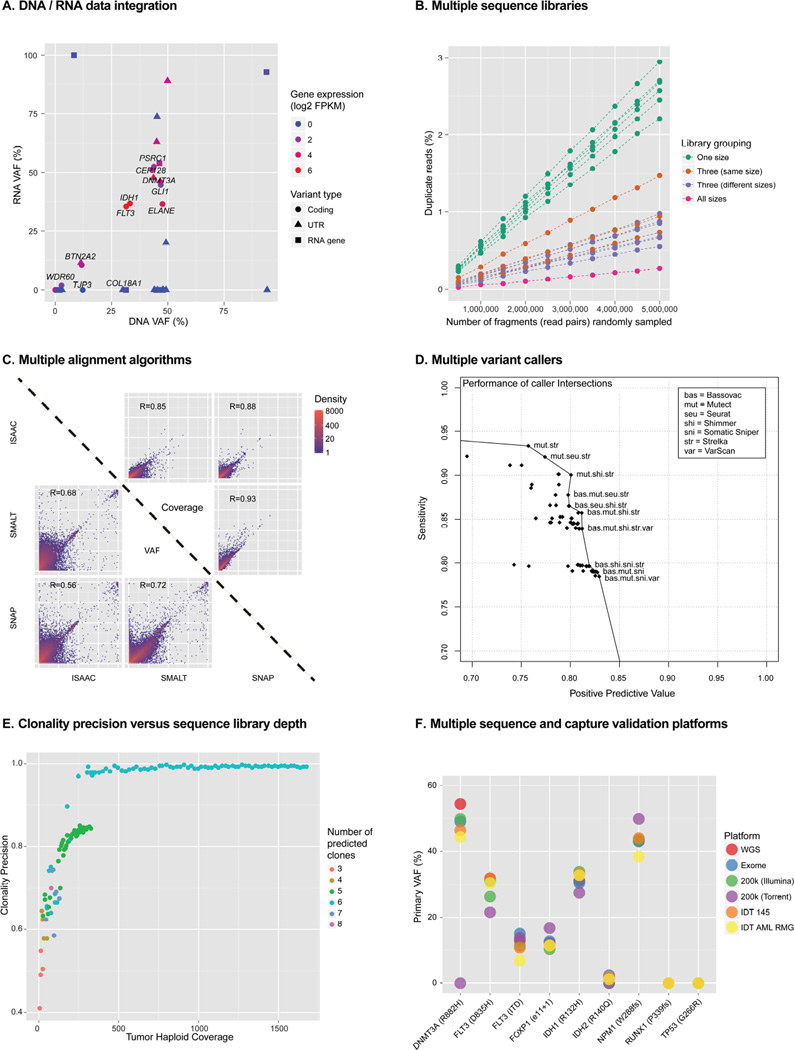
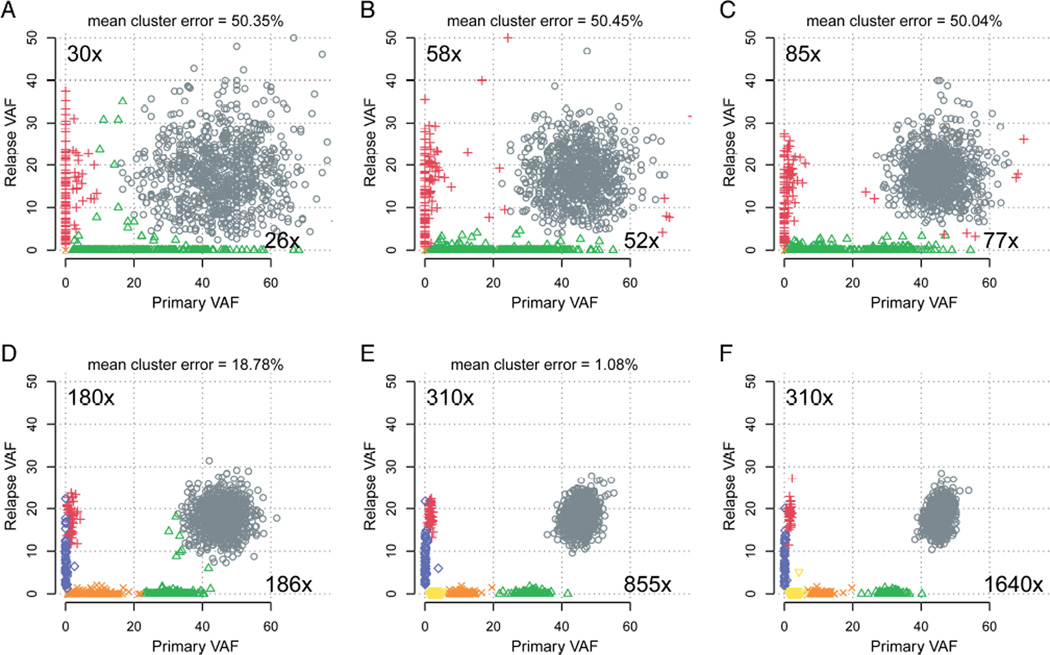
Tumors are typically sequenced to depths of 75-100× (exome) or 30-50× (whole genome). We demonstrate that current sequencing paradigms are inadequate for tumors that are impure, aneuploid or clonally heterogeneous. To reassess optimal sequencing strategies, we performed ultra-deep (up to ~312×) whole genome sequencing (WGS) and exome capture (up to ~433×) of a primary acute myeloid leukemia, its subsequent relapse, and a matched normal skin sample. We tested multiple alignment and variant calling algorithms and validated ~200,000 putative SNVs by sequencing them to depths of ~1,000×. Additional targeted sequencing provided over 10,000× coverage and ddPCR assays provided up to ~250,000× sampling of selected sites. We evaluated the effects of different library generation approaches, depth of sequencing, and analysis strategies on the ability to effectively characterize a complex tumor. This dataset, representing the most comprehensively sequenced tumor described to date, will serve as an invaluable community resource (dbGaP accession id phs000159).
Figures
References
-
- Borad MJ, Champion MD, Egan JB, Liang WS, Fonseca R, Bryce AH, McCullough AE, Barrett MT, Hunt K, Patel MD, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014;10:e1004135. - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
