

The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome
- PMID: 26647311
- PMCID: PMC4731022
- DOI: 10.1093/hmg/ddv496
The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome
Abstract
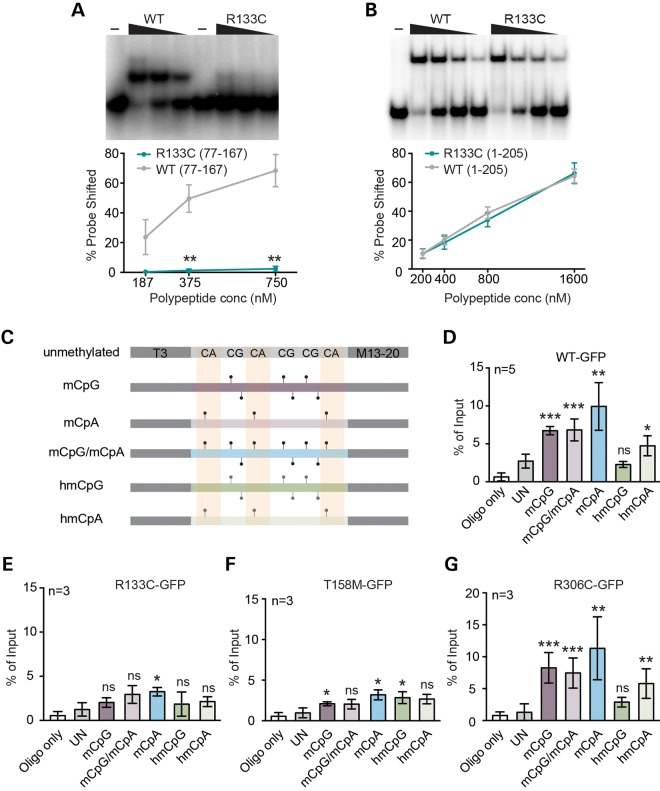
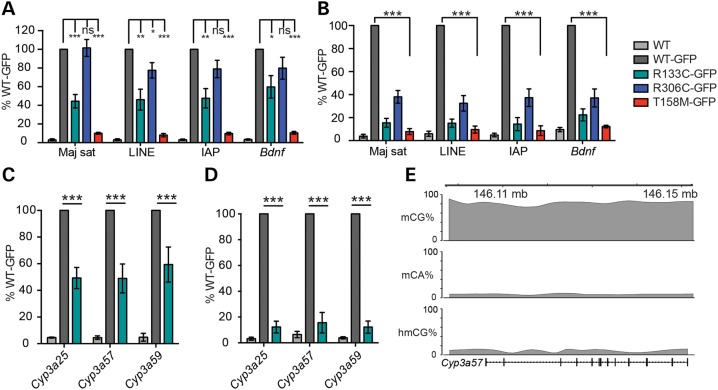
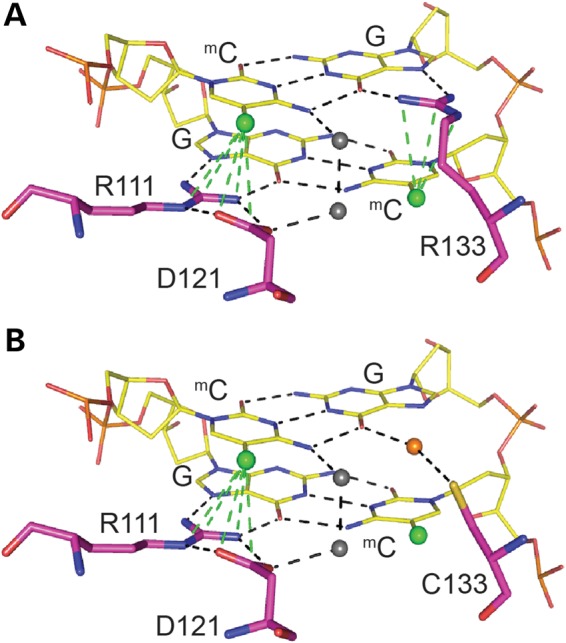
Rett syndrome is caused by mutations in the X-linked MECP2 gene, which encodes a chromosomal protein that binds to methylated DNA. Mouse models mirror the human disorder and therefore allow investigation of phenotypes at a molecular level. We describe an Mecp2 allelic series representing the three most common missense Rett syndrome (RTT) mutations, including first reports of Mecp2[R133C] and Mecp2[T158M] knock-in mice, in addition to Mecp2[R306C] mutant mice. Together these three alleles comprise ∼25% of all RTT mutations in humans, but they vary significantly in average severity. This spectrum is mimicked in the mouse models; R133C being least severe, T158M most severe and R306C of intermediate severity. Both R133C and T158M mutations cause compound phenotypes at the molecular level, combining compromised DNA binding with reduced stability, the destabilizing effect of T158M being more severe. Our findings contradict the hypothesis that the R133C mutation exclusively abolishes binding to hydroxymethylated DNA, as interactions with DNA containing methyl-CG, methyl-CA and hydroxymethyl-CA are all reduced in vivo. We find that MeCP2[T158M] is significantly less stable than MeCP2[R133C], which may account for the divergent clinical impact of the mutations. Overall, this allelic series recapitulates human RTT severity, reveals compound molecular aetiologies and provides a valuable resource in the search for personalized therapeutic interventions.
© The Author 2015. Published by Oxford University Press.
Figures
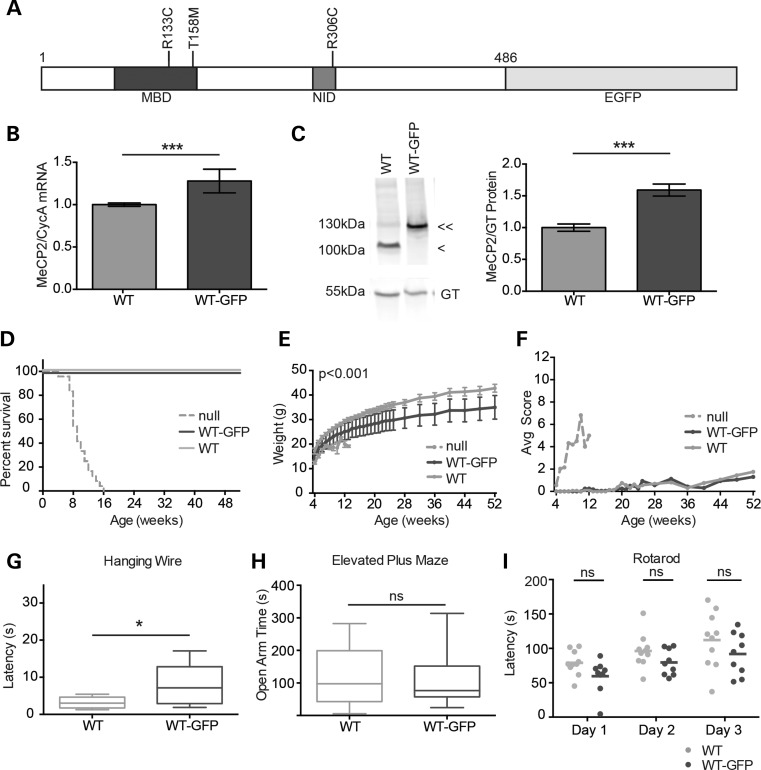
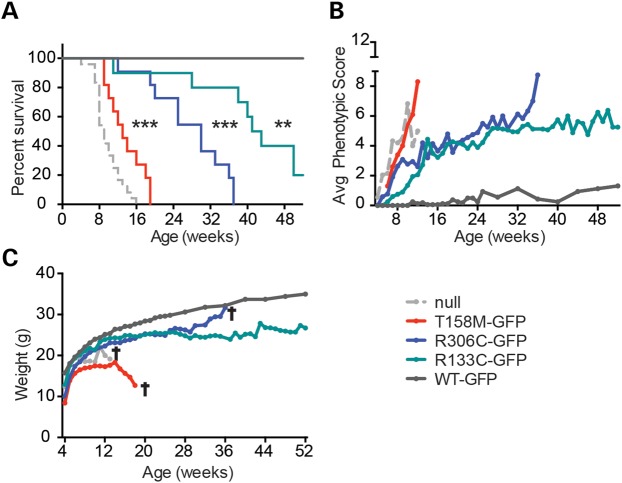
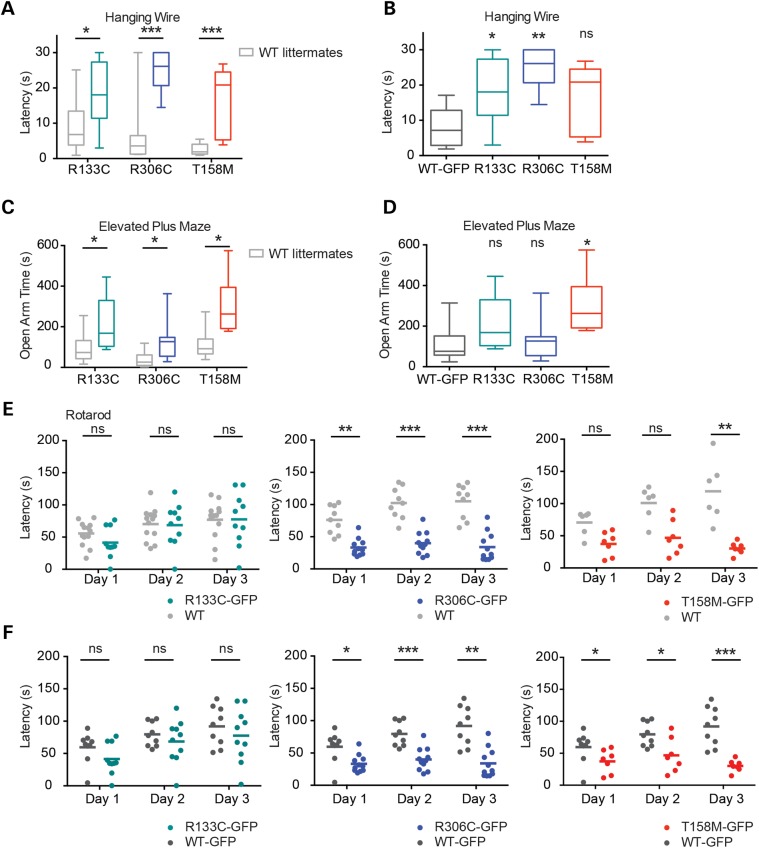
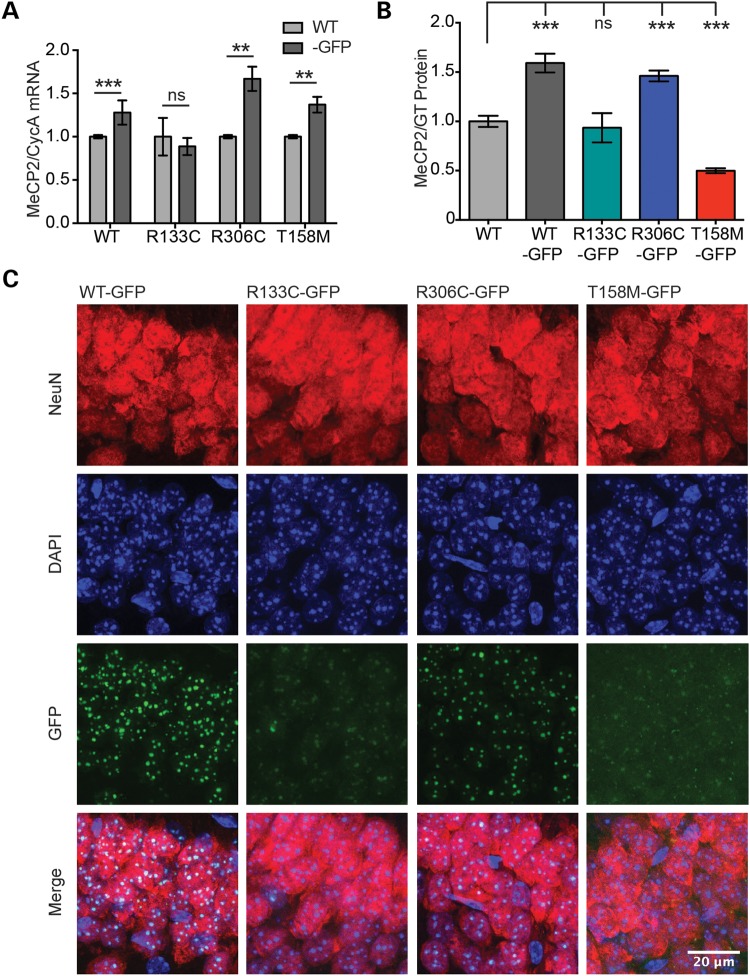
References
-
- Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genet., 23, 185–188. - PubMed
-
- Guy J., Hendrich B., Holmes M., Martin J.E., Bird A. (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet, 27, 322–326. - PubMed
-
- Chen R.Z., Akbarian S., Tudor M., Jaenisch R. (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet, 27, 327–331. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
