

Sanfilippo syndrome: causes, consequences, and treatments
- PMID: 26648750
- PMCID: PMC4664539
- DOI: 10.2147/TACG.S57672
Sanfilippo syndrome: causes, consequences, and treatments
Abstract
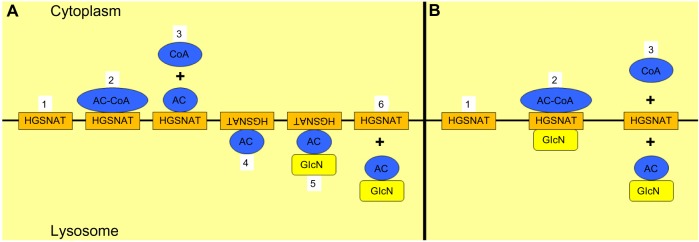
Sanfilippo syndrome, or mucopolysaccharidosis (MPS) type III, refers to one of five autosomal recessive, neurodegenerative lysosomal storage disorders (MPS IIIA to MPS IIIE) whose symptoms are caused by the deficiency of enzymes involved exclusively in heparan sulfate degradation. The primary characteristic of MPS III is the degeneration of the central nervous system, resulting in mental retardation and hyperactivity, typically commencing during childhood. The significance of the order of events leading from heparan sulfate accumulation through to downstream changes in the levels of biomolecules within the cell and ultimately the (predominantly neuropathological) clinical symptoms is not well understood. The genes whose deficiencies cause the MPS III subtypes have been identified, and their gene products, as well as a selection of disease-causing mutations, have been characterized to varying degrees with respect to both frequency and direct biochemical consequences. A number of genetic and biochemical diagnostic methods have been developed and adopted by diagnostic laboratories. However, there is no effective therapy available for any form of MPS III, with treatment currently limited to clinical management of neurological symptoms. The availability of animal models for all forms of MPS III, whether spontaneous or generated via gene targeting, has contributed to improved understanding of the MPS III subtypes, and has provided and will deliver invaluable tools to appraise emerging therapies. Indeed, clinical trials to evaluate intrathecally-delivered enzyme replacement therapy in MPS IIIA patients, and gene therapy for MPS IIIA and MPS IIIB patients are planned or underway.
Keywords: Sanfilippo syndrome; lysosomal storage disease; mucopolysaccharidosis III.
Figures
References
-
- Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–486. - PubMed
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. - PubMed
-
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3421–3452.
-
- Kresse H. Mucopolysaccharidosis 3 A (Sanfilippo A disease): deficiency of a heparin sulfamidase in skin fibroblasts and leucocytes. Biochem Biophys Res Commun. 1973;54:1111–1118. - PubMed
-
- von Figura K. Human α-N-acetylglucosaminidase. 1. Purification and properties. Eur J Biochem. 1977;80:523–533. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
