

Cholesterol Modifies Huntingtin Binding to, Disruption of, and Aggregation on Lipid Membranes
- PMID: 26652744
- PMCID: PMC4956082
- DOI: 10.1021/acs.biochem.5b00900
Cholesterol Modifies Huntingtin Binding to, Disruption of, and Aggregation on Lipid Membranes
Abstract
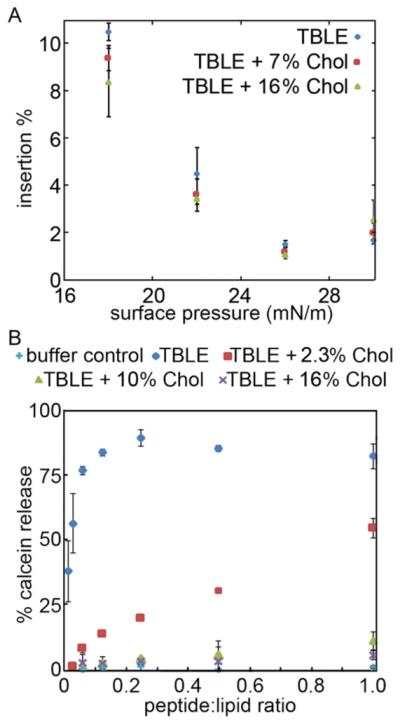
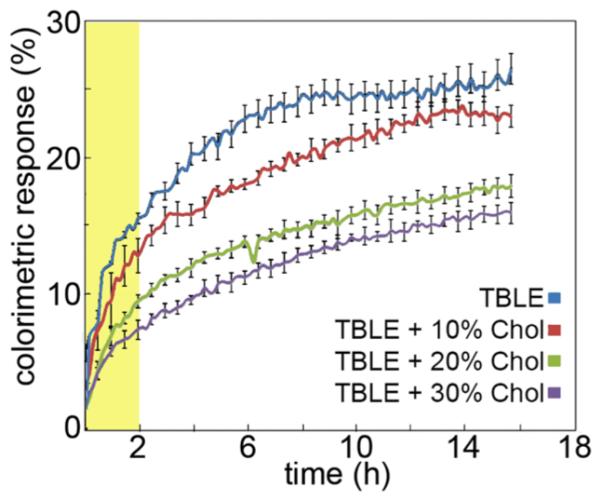
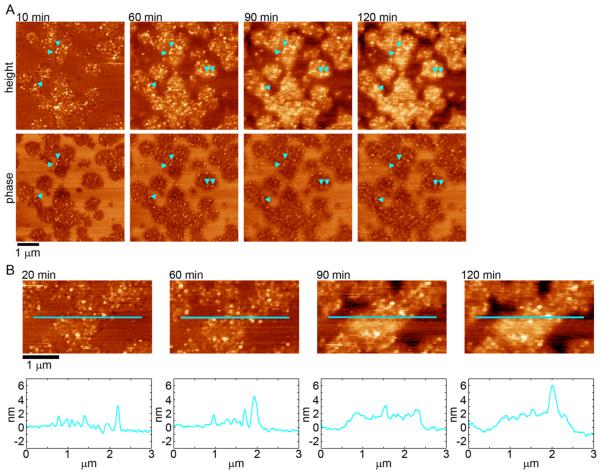
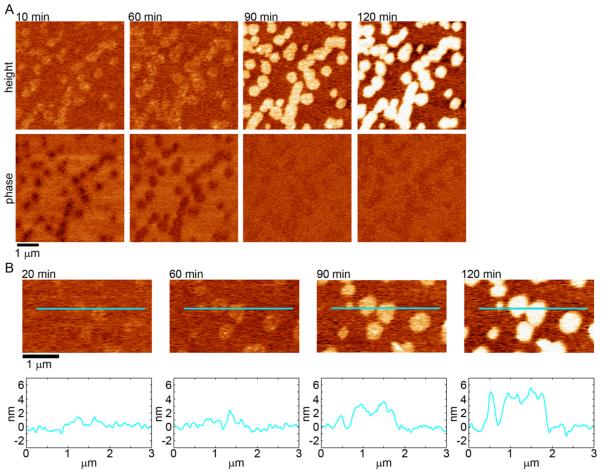
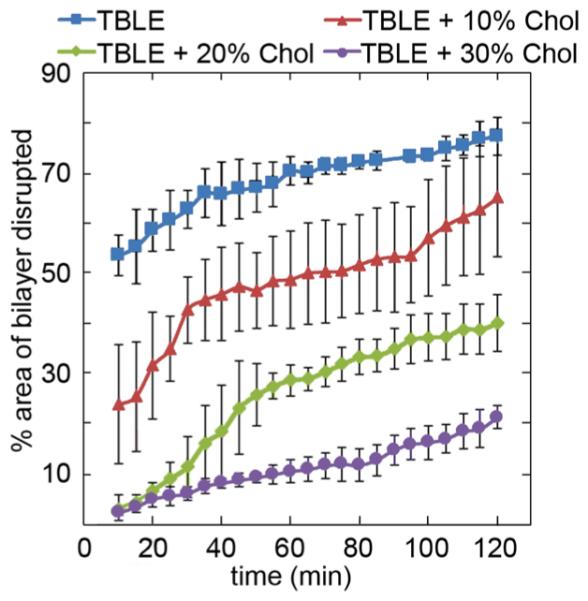
Huntington's disease (HD) is an inherited neurodegenerative disease caused by abnormally long CAG-repeats in the huntingtin gene that encode an expanded polyglutamine (polyQ) domain near the N-terminus of the huntingtin (htt) protein. Expanded polyQ domains are directly correlated to disease-related htt aggregation. Htt is found highly associated with a variety of cellular and subcellular membranes that are predominantly comprised of lipids. Since cholesterol homeostasis is altered in HD, we investigated how varying cholesterol content modifies the interactions between htt and lipid membranes. A combination of Langmuir trough monolayer techniques, vesicle permeability and binding assays, and in situ atomic force microscopy were used to directly monitor the interaction of a model, synthetic htt peptide and a full-length htt-exon1 recombinant protein with model membranes comprised of total brain lipid extract (TBLE) and varying amounts of exogenously added cholesterol. As the cholesterol content of the membrane increased, the extent of htt insertion decreased. Vesicles containing extra cholesterol were resistant to htt-induced permeabilization. Morphological and mechanical changes in the bilayer associated with exposure to htt were also drastically altered by the presence of cholesterol. Disrupted regions of pure TBLE bilayers were grainy in appearance and associated with a large number of globular aggregates. In contrast, morphological changes induced by htt in bilayers enriched in cholesterol were plateau-like with a smooth appearance. Collectively, these observations suggest that the presence and amount of cholesterol in lipid membranes play a critical role in htt binding and aggregation on lipid membranes.
Figures
References
-
- The Huntington Disease Collaborative Research Group. MacDonald ME, et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell. 1993;72:971–983. - PubMed
-
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. - PubMed
-
- Ravina B, Romer M, Constantinescu R, Biglan K, Brocht A, Kieburtz K, Shoulson I, McDermott MP. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov. Disord. 2008;23:1223–1227. - PubMed
-
- Kim YJ, Yi Y, Sapp E, Wang YM, Cuiffo B, Kegel KB, Qin ZH, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. U. S. A. 2001;98:12784–12789. - PMC - PubMed
-
- Ratovitski T, Gucek M, Jiang H, Chighladze E, Waldron E, D’Ambola J, Hou Z, Liang Y, Poirier MA, Hirschhorn RR, Graham R, Hayden MR, Cole RN, Ross CA. Mutant Huntingtin N-terminal Fragments of Specific Size Mediate Aggregation and Toxicity in Neuronal Cells. J. Biol. Chem. 2009;284:10855–10867. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
