

Oligodendrocyte death results in immune-mediated CNS demyelination
- PMID: 26656646
- PMCID: PMC4837900
- DOI: 10.1038/nn.4193
Oligodendrocyte death results in immune-mediated CNS demyelination
Abstract
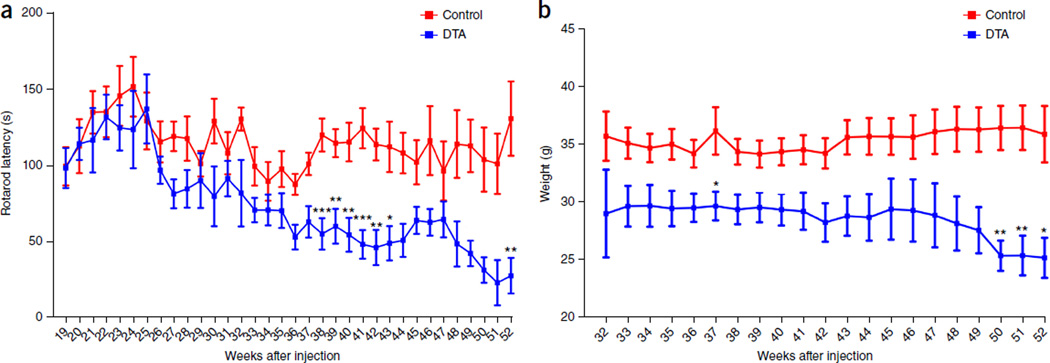
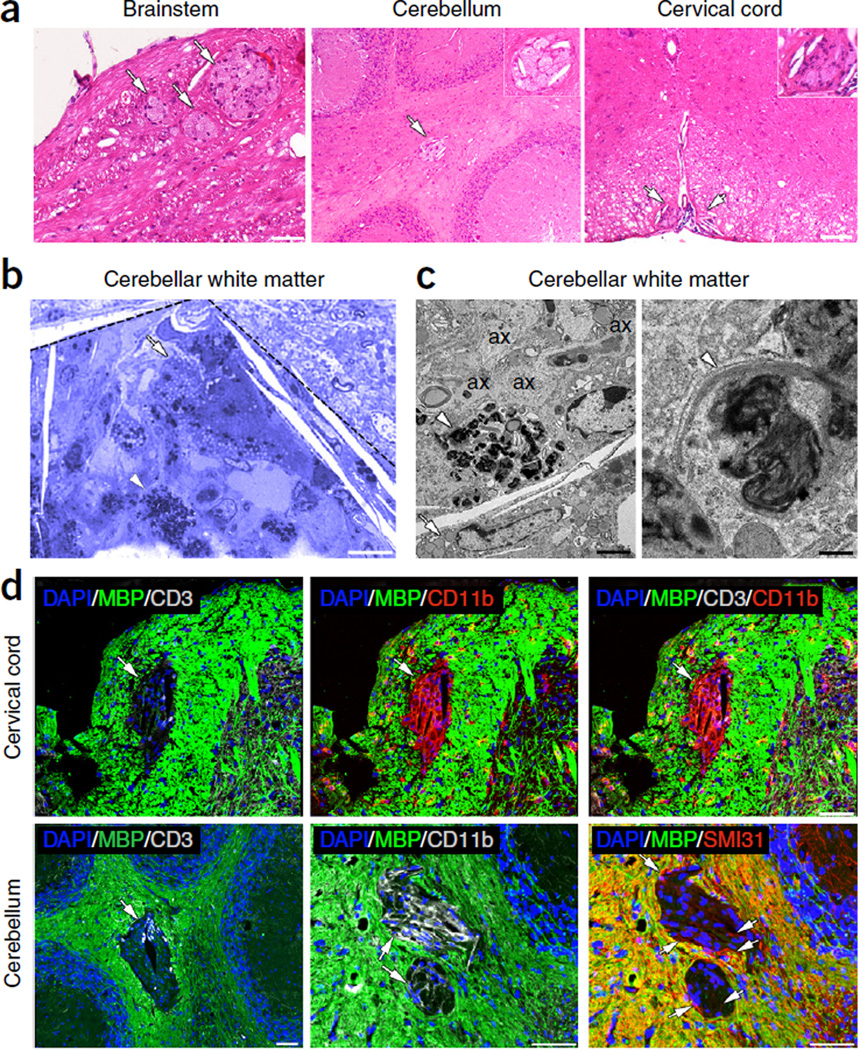
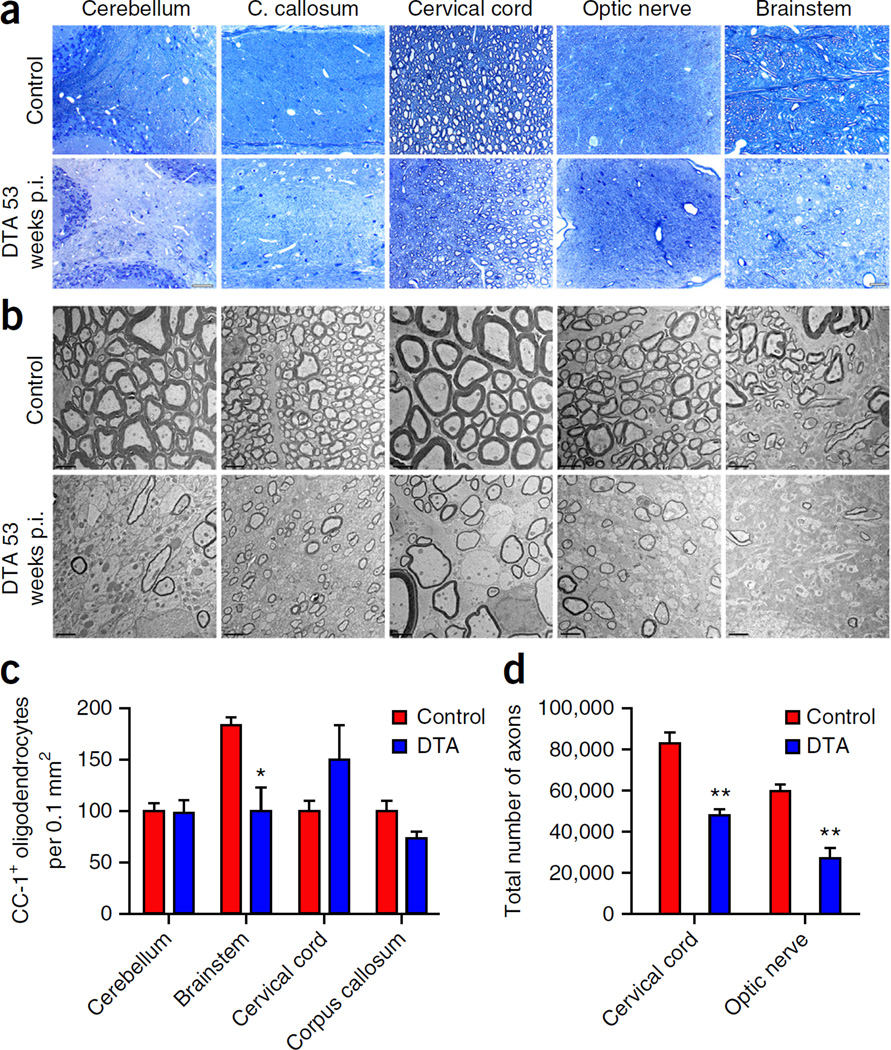
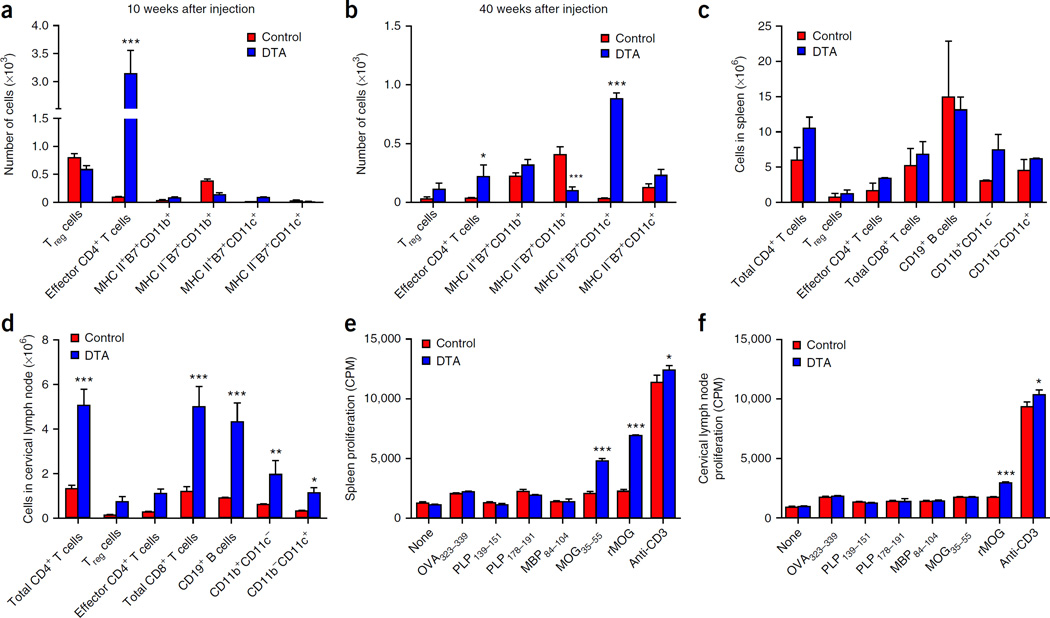
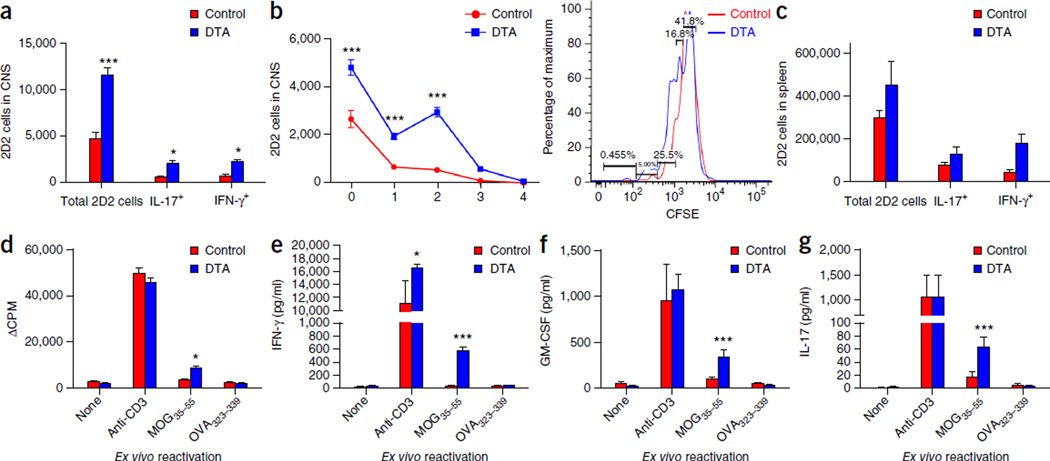
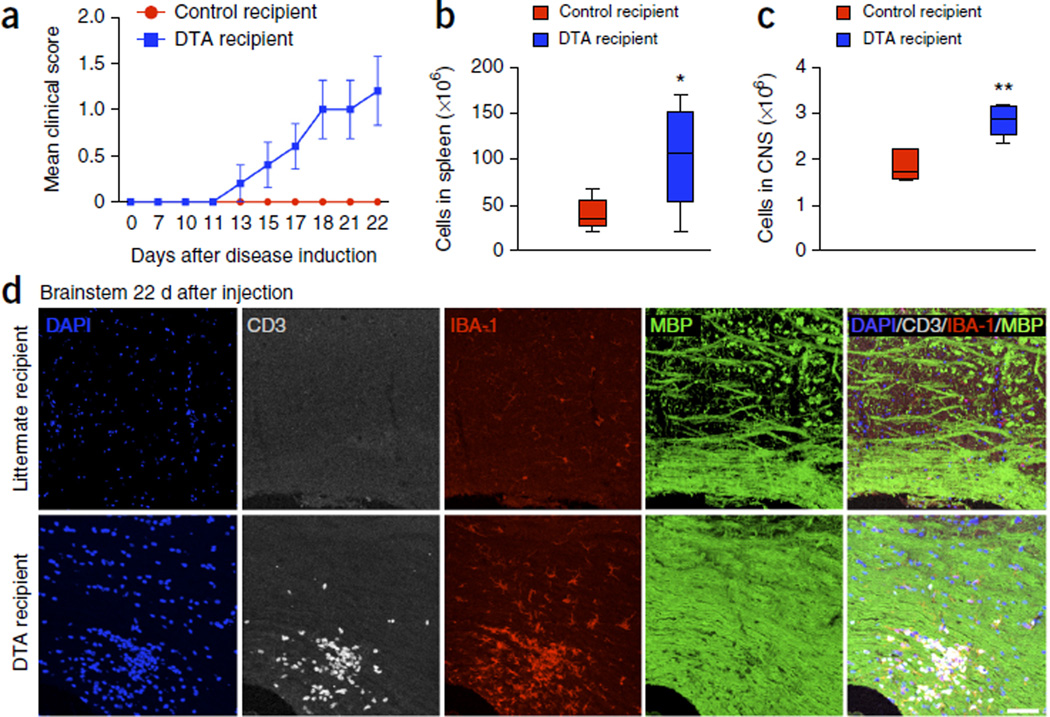
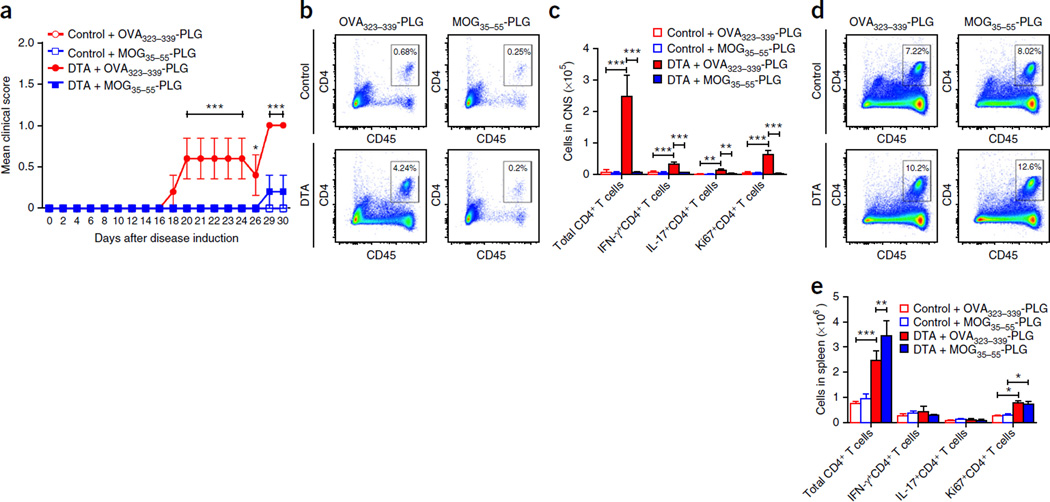
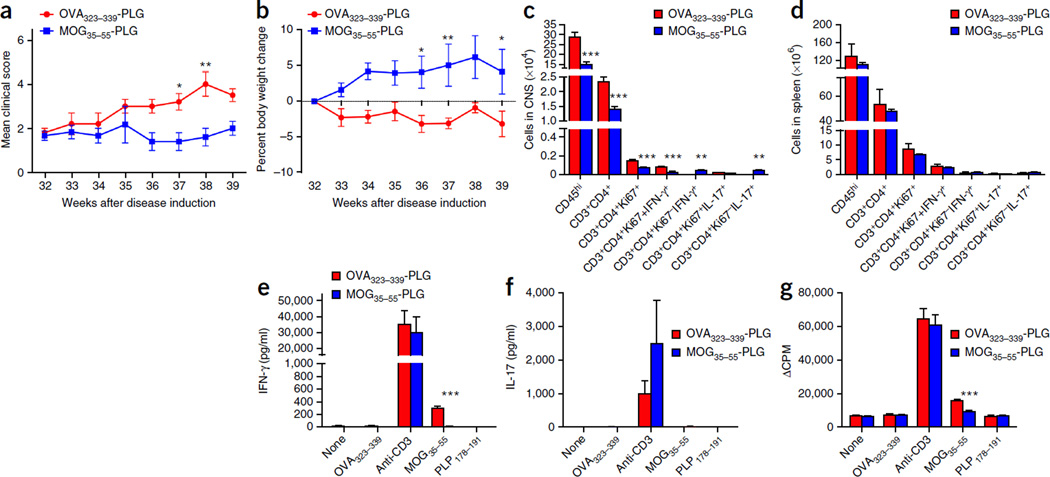
Although multiple sclerosis is a common neurological disorder, the origin of the autoimmune response against myelin, which is the characteristic feature of the disease, remains unclear. To investigate whether oligodendrocyte death could cause this autoimmune response, we examined the oligodendrocyte ablation Plp1-CreER(T);ROSA26-eGFP-DTA (DTA) mouse model. Approximately 30 weeks after recovering from oligodendrocyte loss and demyelination, DTA mice develop a fatal secondary disease characterized by extensive myelin and axonal loss. Strikingly, late-onset disease was associated with increased numbers of T lymphocytes in the CNS and myelin oligodendrocyte glycoprotein (MOG)-specific T cells in lymphoid organs. Transfer of T cells derived from DTA mice to naive recipients resulted in neurological defects that correlated with CNS white matter inflammation. Furthermore, immune tolerization against MOG ameliorated symptoms. Overall, these data indicate that oligodendrocyte death is sufficient to trigger an adaptive autoimmune response against myelin, suggesting that a similar process can occur in the pathogenesis of multiple sclerosis.
Figures
Comment in
-
Neurological disorders: A second wave.Nat Rev Neurosci. 2016 Feb;17(2):76. doi: 10.1038/nrn.2016.5. Nat Rev Neurosci. 2016. PMID: 26806625 No abstract available.
References
-
- Nakahara J, Aiso S, Suzuki N. Autoimmune versus oligodendrogliopathy: the pathogenesis of multiple sclerosis. Arch. Immunol. Ther. Exp. (Warsz.) 2010;58:325–333. - PubMed
-
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 2007;8:913–919. - PubMed
-
- Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012;13:507–514. - PubMed
-
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 2004;55:458–468. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
