

Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features
- PMID: 26656649
- PMCID: PMC4902791
- DOI: 10.1038/gim.2015.161
Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features
Abstract
Purpose: The pace of Mendelian gene discovery is slowed by the "n-of-1 problem"-the difficulty of establishing the causality of a putatively pathogenic variant in a single person or family. Identification of an unrelated person with an overlapping phenotype and suspected pathogenic variant in the same gene can overcome this barrier, but it is often impeded by lack of a convenient or widely available way to share data on candidate variants/genes among families, clinicians, and researchers.
Methods: Social networking among families, clinicians, and researchers was used to identify three children with variants of unknown significance in KDM1A and similar phenotypes.
Results: De novo variants in KDM1A underlie a new syndrome characterized by developmental delay and distinctive facial features.
Conclusion: Social networking is a potentially powerful strategy to discover genes for rare Mendelian conditions, particularly those with nonspecific phenotypic features. To facilitate the efforts of families to share phenotypic and genomic information with each other, clinicians, and researchers, we developed the Repository for Mendelian Genomics Family Portal (RMD-FP; http://uwcmg.org/#/family). Design and development of MyGene2 (http://www.mygene2.org), a Web-based tool that enables families, clinicians, and researchers to search for gene matches based on analysis of phenotype and exome data deposited into the RMD-FP, is under way.Genet Med 18 8, 788-795.
Conflict of interest statement
M.J.B., H.K.T., and J-H.Y. have a patent application pending on My46. The other authors declare no conflict of interest.
Figures
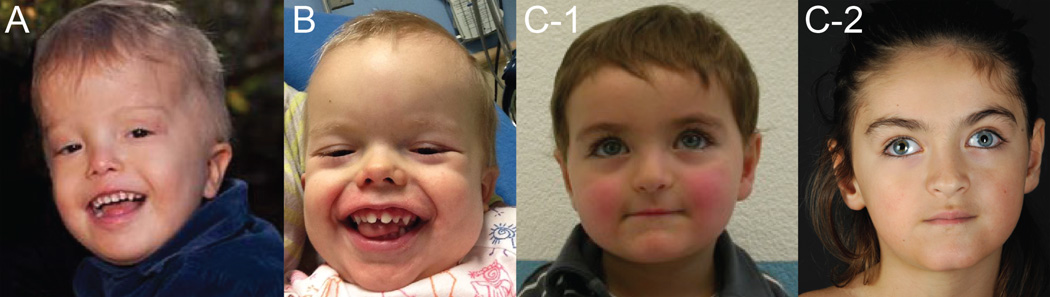
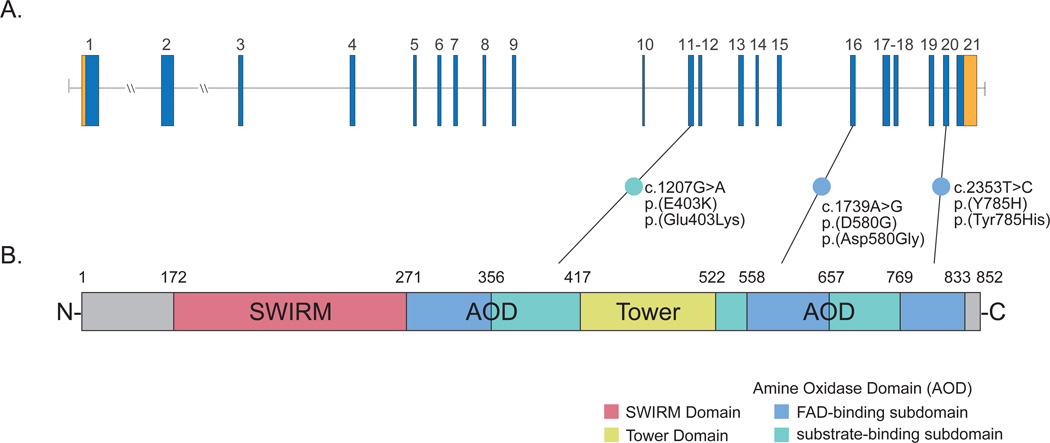

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
