

Fine-mapping cellular QTLs with RASQUAL and ATAC-seq
- PMID: 26656845
- PMCID: PMC5098600
- DOI: 10.1038/ng.3467
Fine-mapping cellular QTLs with RASQUAL and ATAC-seq
Erratum in
-
Corrigendum: Fine-mapping cellular QTLs with RASQUAL and ATAC-seq.Nat Genet. 2016 Apr;48(4):473. doi: 10.1038/ng0329-473e. Nat Genet. 2016. PMID: 27023776 No abstract available.
Abstract
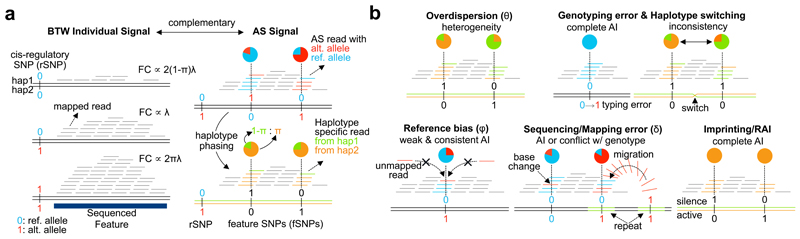
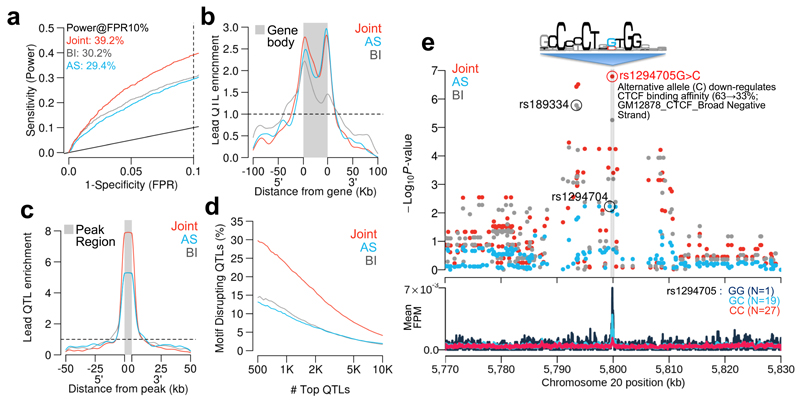
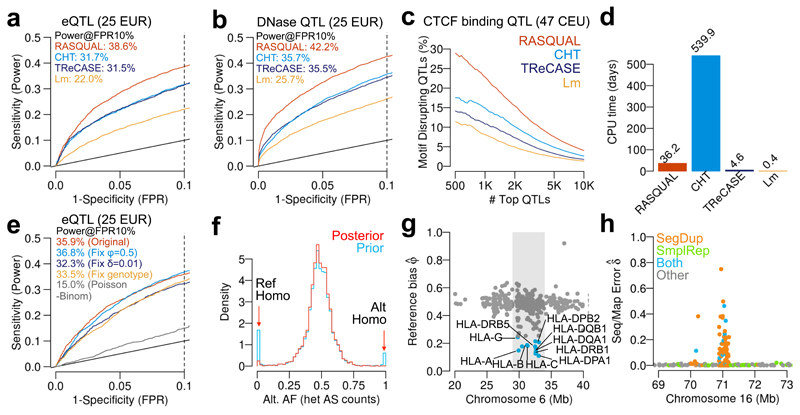
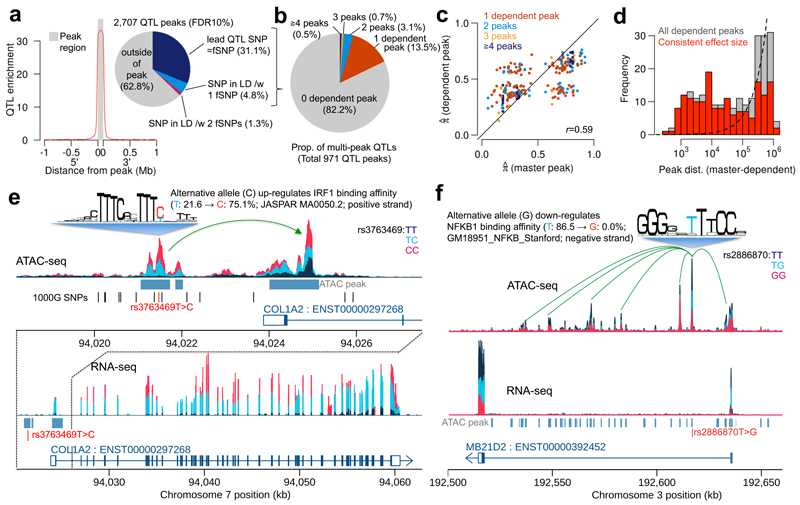
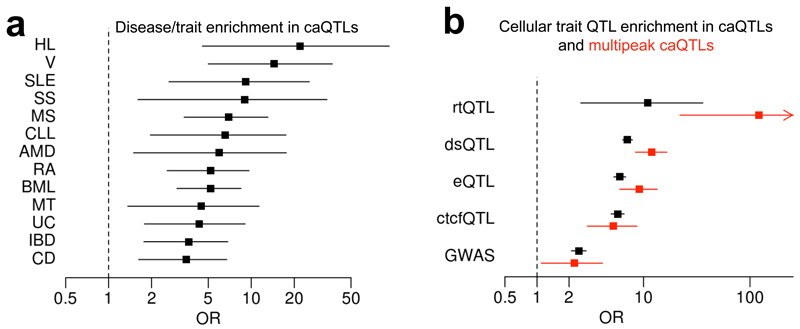
When cellular traits are measured using high-throughput DNA sequencing, quantitative trait loci (QTLs) manifest as fragment count differences between individuals and allelic differences within individuals. We present RASQUAL (Robust Allele-Specific Quantitation and Quality Control), a new statistical approach for association mapping that models genetic effects and accounts for biases in sequencing data using a single, probabilistic framework. RASQUAL substantially improves fine-mapping accuracy and sensitivity relative to existing methods in RNA-seq, DNase-seq and ChIP-seq data. We illustrate how RASQUAL can be used to maximize association detection by generating the first map of chromatin accessibility QTLs (caQTLs) in a European population using ATAC-seq. Despite a modest sample size, we identified 2,707 independent caQTLs (at a false discovery rate of 10%) and demonstrated how RASQUAL and ATAC-seq can provide powerful information for fine-mapping gene-regulatory variants and for linking distal regulatory elements with gene promoters. Our results highlight how combining between-individual and allele-specific genetic signals improves the functional interpretation of noncoding variation.
Conflict of interest statement
Conflicts of Interest: None declared
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
