

Tumor suppressor BTG1 promotes PRMT1-mediated ATF4 function in response to cellular stress
- PMID: 26657730
- PMCID: PMC4823095
- DOI: 10.18632/oncotarget.6519
Tumor suppressor BTG1 promotes PRMT1-mediated ATF4 function in response to cellular stress
Abstract
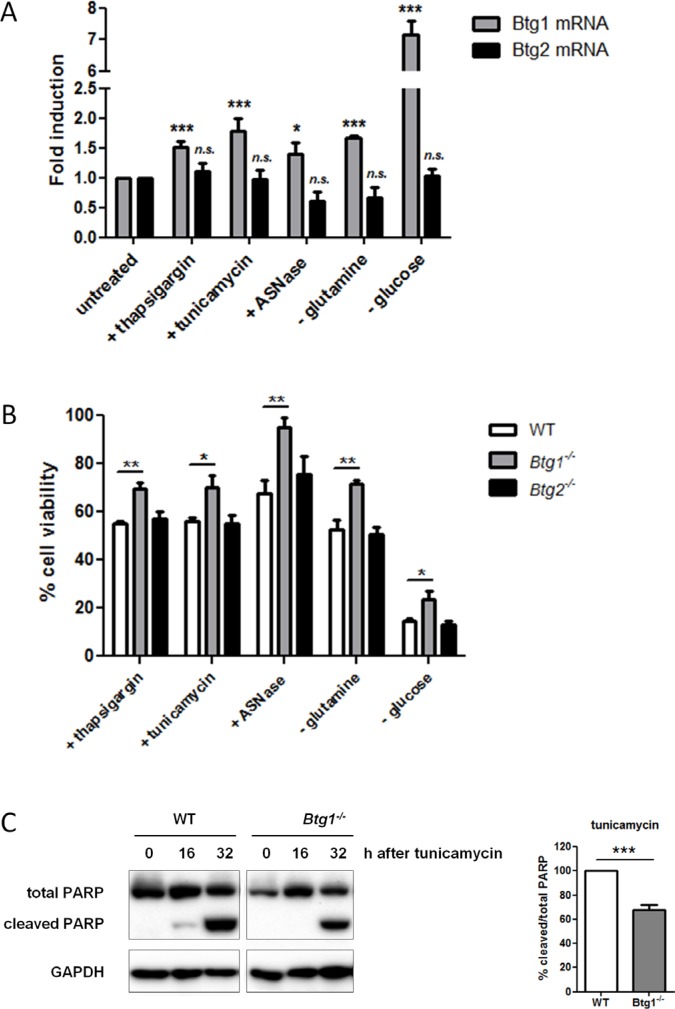
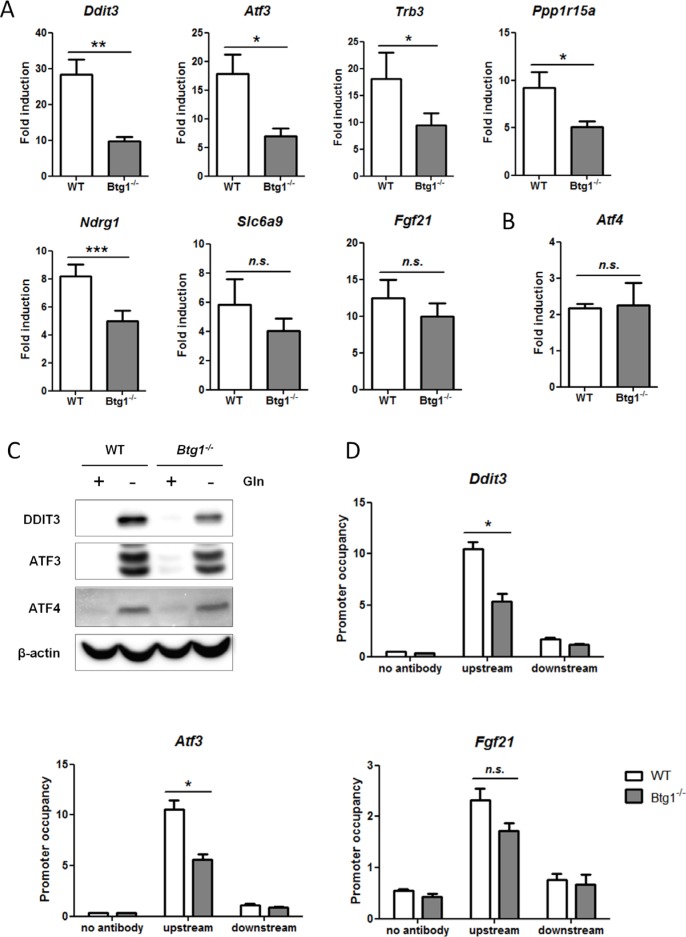
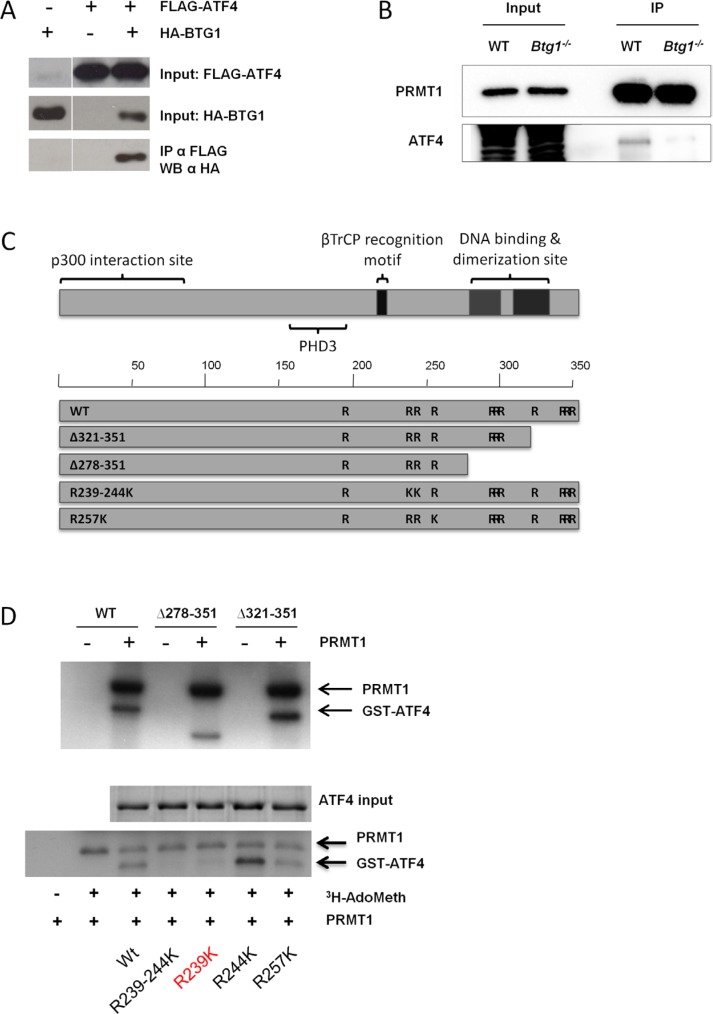
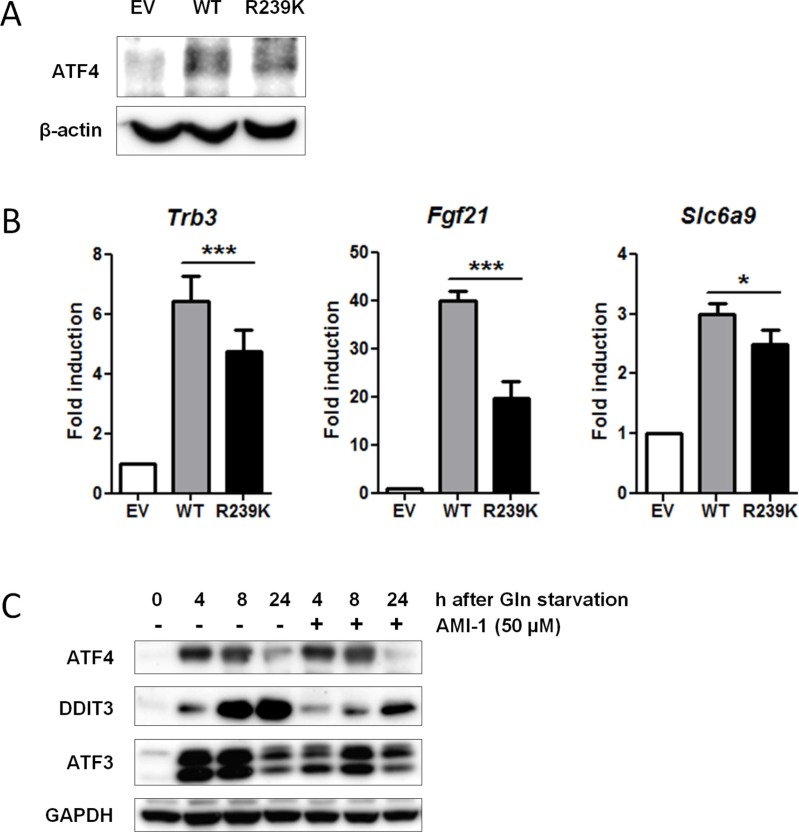
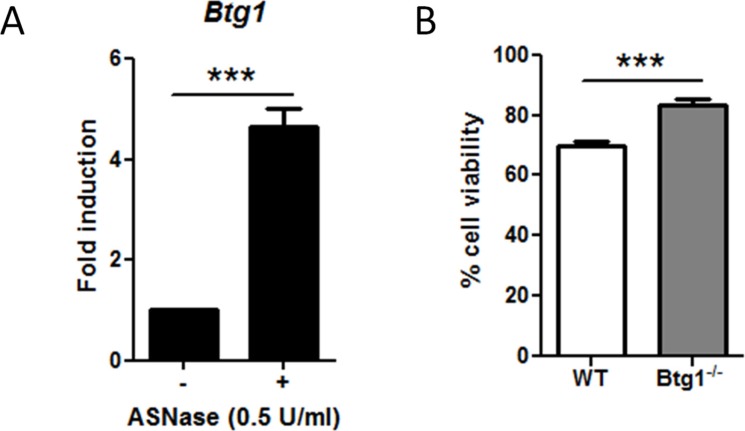
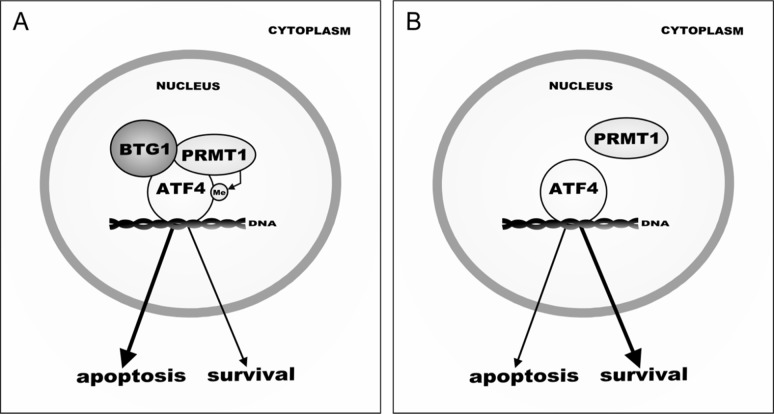
Cancer cells are frequently exposed to physiological stress conditions such as hypoxia and nutrient limitation. Escape from stress-induced apoptosis is one of the mechanisms used by malignant cells to survive unfavorable conditions. B-cell Translocation Gene 1 (BTG1) is a tumor suppressor that is frequently deleted in acute lymphoblastic leukemia and recurrently mutated in diffuse large B cell lymphoma. Moreover, low BTG1 expression levels have been linked to poor outcome in several solid tumors. How loss of BTG1 function contributes to tumor progression is not well understood. Here, using Btg1 knockout mice, we demonstrate that loss of Btg1 provides a survival advantage to primary mouse embryonic fibroblasts (MEFs) under stress conditions. This pro-survival effect involves regulation of Activating Transcription Factor 4 (ATF4), a key mediator of cellular stress responses. We show that BTG1 interacts with ATF4 and positively modulates its activity by recruiting the protein arginine methyl transferase PRMT1 to methylate ATF4 on arginine residue 239. We further extend these findings to B-cell progenitors, by showing that loss of Btg1 expression enhances stress adaptation of mouse bone marrow-derived B cell progenitors. In conclusion, we have identified the BTG1/PRMT1 complex as a new modifier of ATF4 mediated stress responses.
Keywords: ATF4; BTG1; PRMT1; cellular stress; leukemia.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Siu F, Bain PJ, LeBlanc-Chaffin R, Chen H, Kilberg MS. ATF4 is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J Biol Chem. 2002;277:24120–24127. - PubMed
-
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29:4424–4435. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
