

The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis
- PMID: 26658360
- PMCID: PMC4677309
- DOI: 10.1038/srep18272
The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis
Abstract
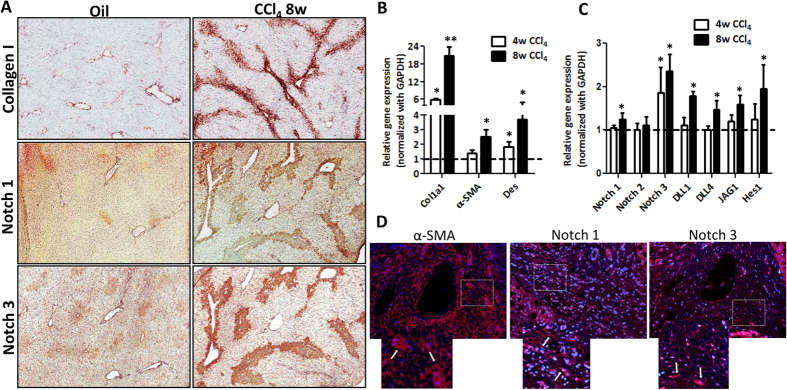
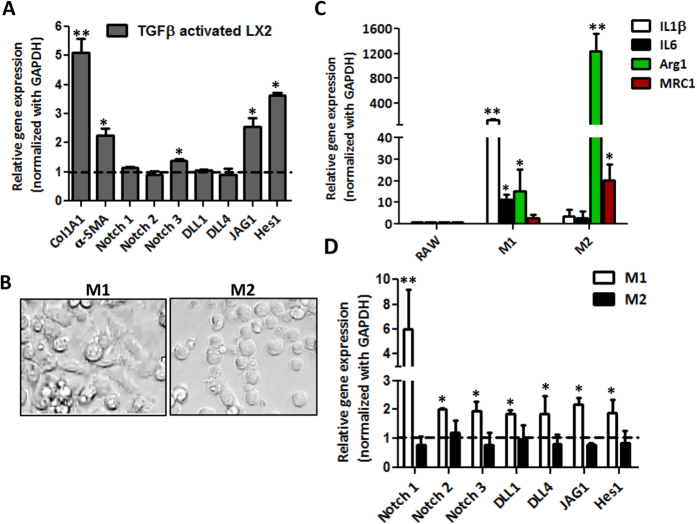
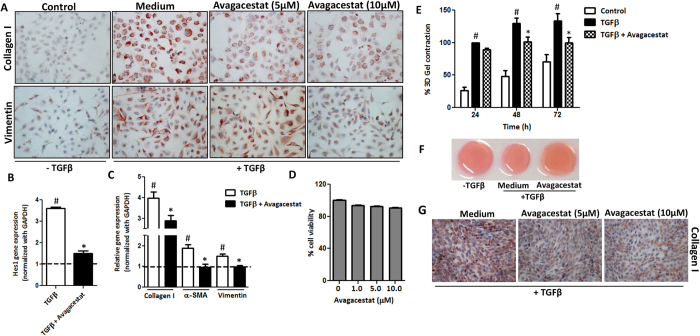
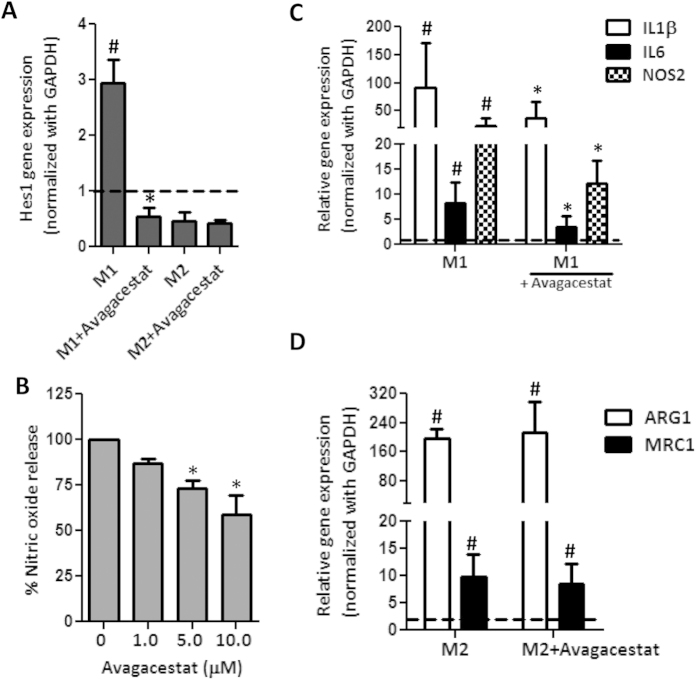
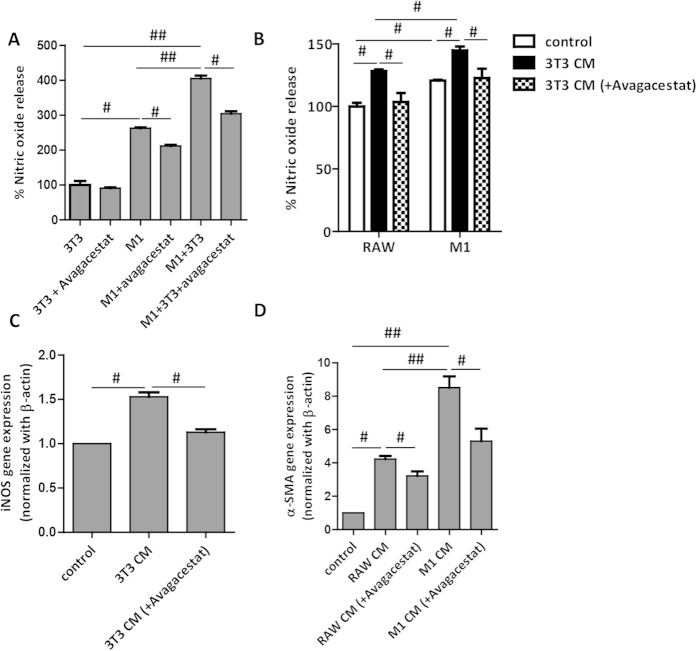
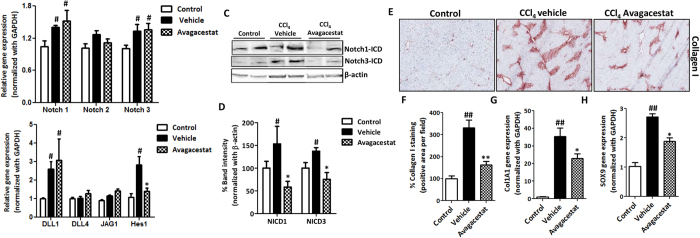
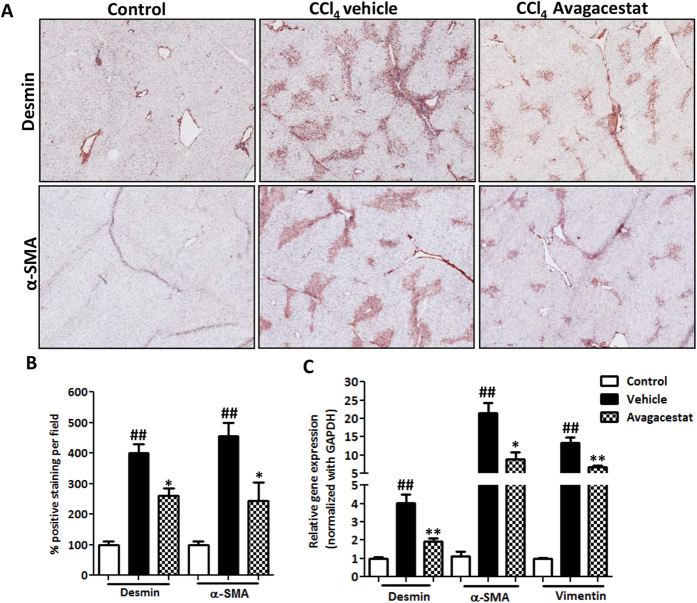
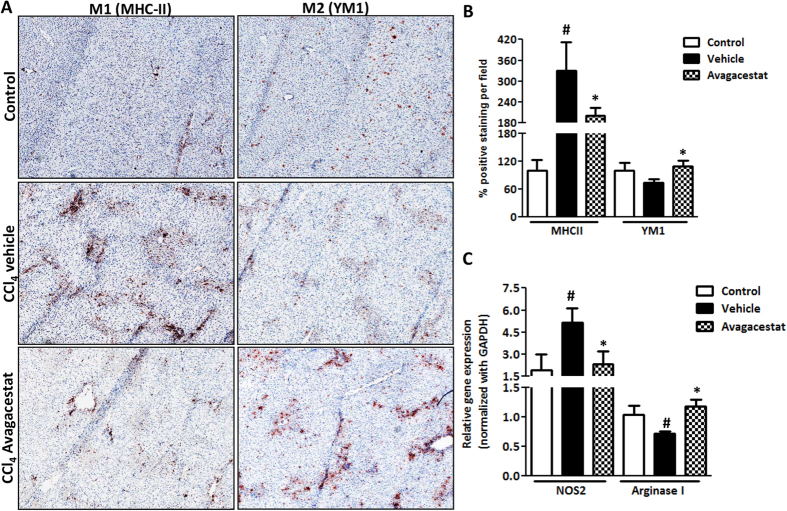
Hepatic stellate cells (HSCs) known as "master producers" and macrophages as "master regulators", are the key cell types that strongly contribute to the progression of liver fibrosis. Since Notch signaling regulates multiple cellular processes, we aimed to study the role of Notch signaling in HSCs differentiation and macrophages polarization and to evaluate its implication in liver fibrogenesis. Notch pathway components were found to be significantly upregulated in TGFβ-activated HSCs, inflammatory M1 macrophages, and in mouse and human fibrotic livers. Interestingly, inhibition of Notch using a selective γ-secretase inhibitor, Avagacestat, significantly inhibited TGFβ-induced HSC activation and contractility, and suppressed M1 macrophages. Additionally, Avagacestat inhibited M1 driven-fibroblasts activation and fibroblasts-driven M1 polarization (nitric oxide release) in fibroblasts and macrophages co-culture, and conditioned medium studies. In vivo, post-disease treatment with Avagacestat significantly attenuated fibrogenesis in CCl4-induced liver fibrosis mouse model. These effects were attributed to the reduction in HSCs activation, and inhibition of inflammatory M1 macrophages and upregulation of suppressive M2 macrophages. These findings suggest that Notch signaling plays a crucial role in HSC activation and M1/M2 polarization of macrophages in liver fibrosis. These results provide new insights for the development of novel therapies against liver fibrosis through modulation of Notch signaling.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
