

Direct Evidence that Myocardial Insulin Resistance following Myocardial Ischemia Contributes to Post-Ischemic Heart Failure
- PMID: 26659007
- PMCID: PMC4677294
- DOI: 10.1038/srep17927
Direct Evidence that Myocardial Insulin Resistance following Myocardial Ischemia Contributes to Post-Ischemic Heart Failure
Abstract
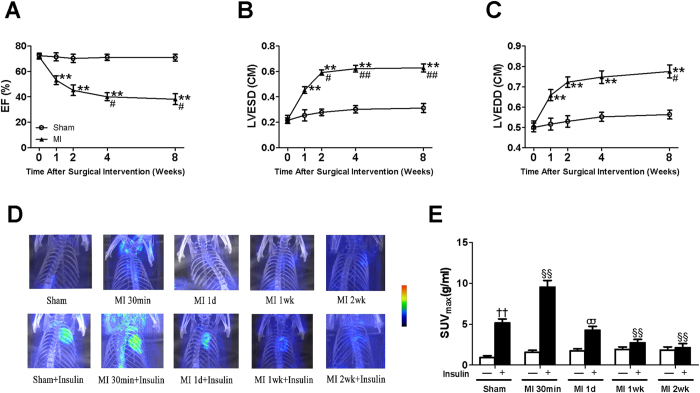
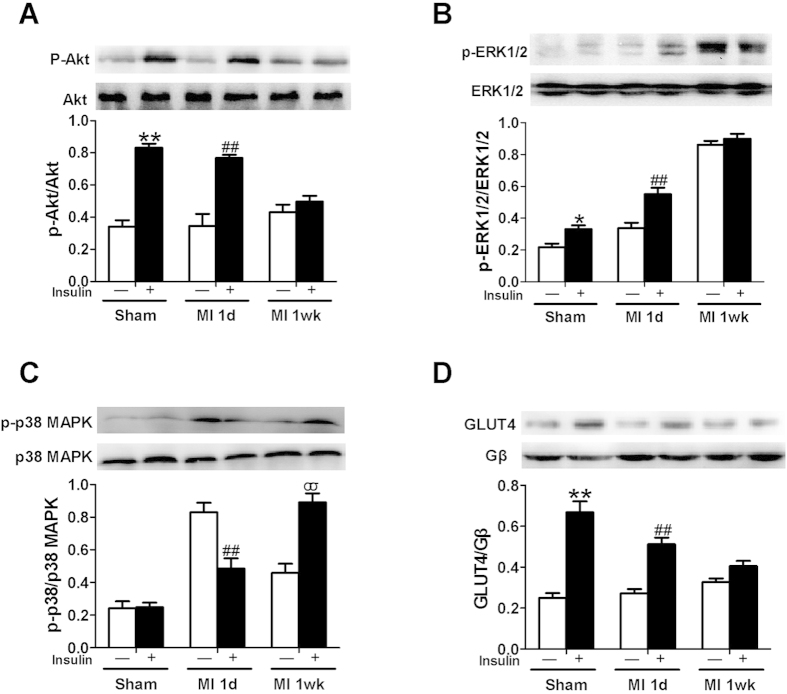
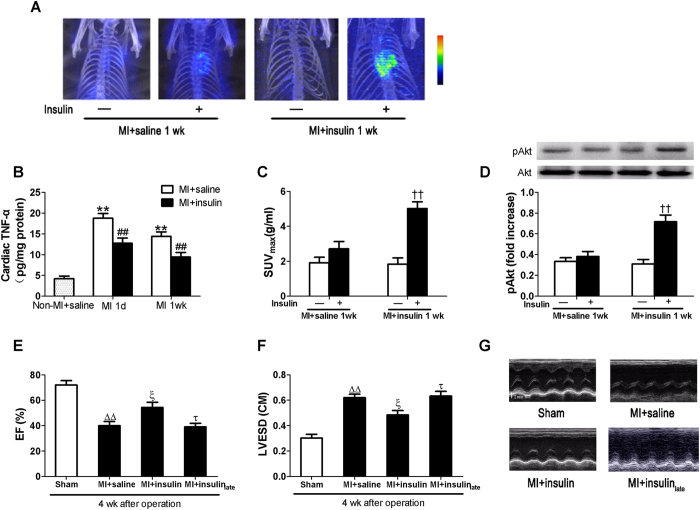
A close link between heart failure (HF) and systemic insulin resistance has been well documented, whereas myocardial insulin resistance and its association with HF are inadequately investigated. This study aims to determine the role of myocardial insulin resistance in ischemic HF and its underlying mechanisms. Male Sprague-Dawley rats subjected to myocardial infarction (MI) developed progressive left ventricular dilation with dysfunction and HF at 4 wk post-MI. Of note, myocardial insulin sensitivity was decreased as early as 1 wk after MI, which was accompanied by increased production of myocardial TNF-α. Overexpression of TNF-α in heart mimicked impaired insulin signaling and cardiac dysfunction leading to HF observed after MI. Treatment of rats with a specific TNF-α inhibitor improved myocardial insulin signaling post-MI. Insulin treatment given immediately following MI suppressed myocardial TNF-α production and improved cardiac insulin sensitivity and opposed cardiac dysfunction/remodeling. Moreover, tamoxifen-induced cardiomyocyte-specific insulin receptor knockout mice exhibited aggravated post-ischemic ventricular remodeling and dysfunction compared with controls. In conclusion, MI induces myocardial insulin resistance (without systemic insulin resistance) mediated partly by ischemia-induced myocardial TNF-α overproduction and promotes the development of HF. Our findings underscore the direct and essential role of myocardial insulin signaling in protection against post-ischemic HF.
Figures



Similar articles
-
Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction.Am J Physiol Heart Circ Physiol. 2016 Nov 1;311(5):H1160-H1169. doi: 10.1152/ajpheart.00114.2016. Epub 2016 Aug 19. Am J Physiol Heart Circ Physiol. 2016. PMID: 27542406
-
Clinical aspects of left ventricular diastolic function assessed by Doppler echocardiography following acute myocardial infarction.Dan Med Bull. 2001 Nov;48(4):199-210. Dan Med Bull. 2001. PMID: 11767125 Review.
-
Ischemia/Infarction.Heart Fail Clin. 2012 Jan;8(1):43-51. doi: 10.1016/j.hfc.2011.08.006. Epub 2011 Oct 14. Heart Fail Clin. 2012. PMID: 22108726 Review.
-
Thoracic spinal cord stimulation improves cardiac contractile function and myocardial oxygen consumption in a porcine model of ischemic heart failure.J Cardiovasc Electrophysiol. 2012 May;23(5):534-40. doi: 10.1111/j.1540-8167.2011.02230.x. Epub 2011 Dec 8. J Cardiovasc Electrophysiol. 2012. PMID: 22151312
-
Combination angiotensin converting enzyme and direct renin inhibition in heart failure following experimental myocardial infarction.Cardiovasc Ther. 2013 Apr;31(2):84-91. doi: 10.1111/j.1755-5922.2011.00292.x. Epub 2011 Jul 4. Cardiovasc Ther. 2013. PMID: 21884026
Cited by
-
Cardiovascular Disease and Exercise: From Molecular Mechanisms to Clinical Applications.J Clin Med. 2022 Dec 19;11(24):7511. doi: 10.3390/jcm11247511. J Clin Med. 2022. Retraction in: J Clin Med. 2023 Mar 09;12(6):2143. doi: 10.3390/jcm12062143. PMID: 36556132 Free PMC article. Retracted. Review.
-
Extracellular matrix remodelling in obesity and metabolic disorders.Life Metab. 2023 Aug;2(4):load021. doi: 10.1093/lifemeta/load021. Epub 2023 May 26. Life Metab. 2023. PMID: 37383542 Free PMC article.
-
Role and mechanism of cardiac insulin resistance in occurrence of heart failure caused by myocardial hypertrophy.Aging (Albany NY). 2019 Aug 28;11(16):6584-6590. doi: 10.18632/aging.102212. Epub 2019 Aug 28. Aging (Albany NY). 2019. PMID: 31461405 Free PMC article.
-
Hippocampus Insulin Receptors Regulate Episodic and Spatial Memory Through Excitatory/Inhibitory Balance.ASN Neuro. 2023 Jan-Dec;15:17590914231206657. doi: 10.1177/17590914231206657. ASN Neuro. 2023. PMID: 37908089 Free PMC article.
-
Fenofibrate Therapy Restores Antioxidant Protection and Improves Myocardial Insulin Resistance in a Rat Model of Metabolic Syndrome and Myocardial Ischemia: The Role of Angiotensin II.Molecules. 2016 Dec 28;22(1):31. doi: 10.3390/molecules22010031. Molecules. 2016. PMID: 28036029 Free PMC article.
References
-
- Kannel W. B., Hjortland M. & Castelli W. P. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 34, 29–34 (1974). - PubMed
-
- Kannel W. B. & McGee D. L. Diabetes and cardiovascular disease. The Framingham study. JAMA 241, 2035–2038 (1979). - PubMed
-
- Fox C. S. et al. Increasing cardiovascular disease burden due to diabetes mellitus: the Framingham Heart Study. Circulation 115, 1544–1550 (2007). - PubMed
-
- Swan J. W. et al. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol 30, 527–532 (1997). - PubMed
-
- AlZadjali M. A. et al. Insulin resistance is highly prevalent and is associated with reduced exercise tolerance in nondiabetic patients with heart failure. J Am Coll Cardiol 53, 747–753 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
