

Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance
- PMID: 26660431
- PMCID: PMC4705608
- DOI: 10.1182/blood-2015-07-604496
Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance
Abstract
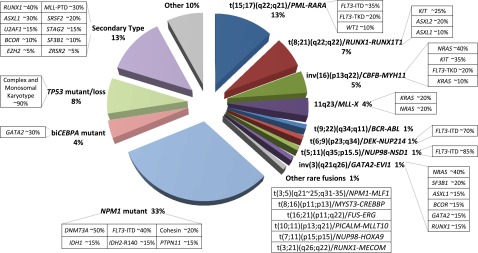
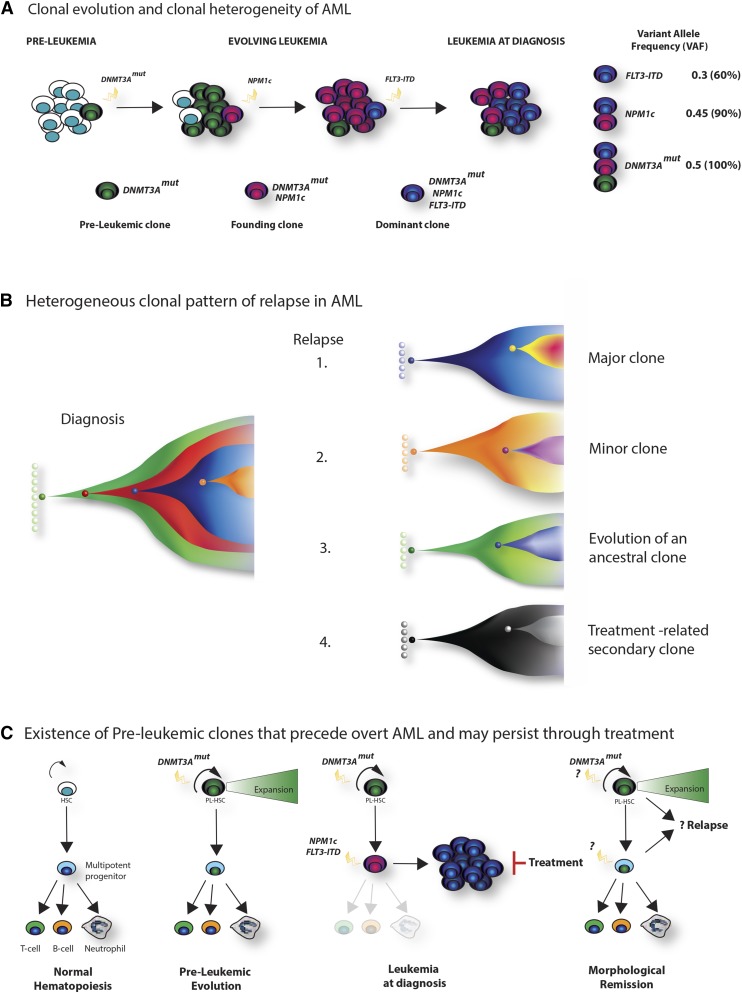
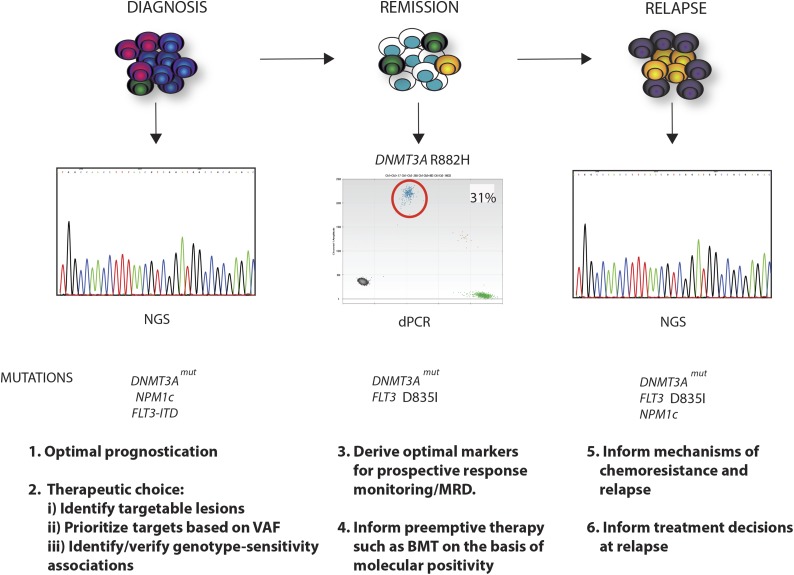
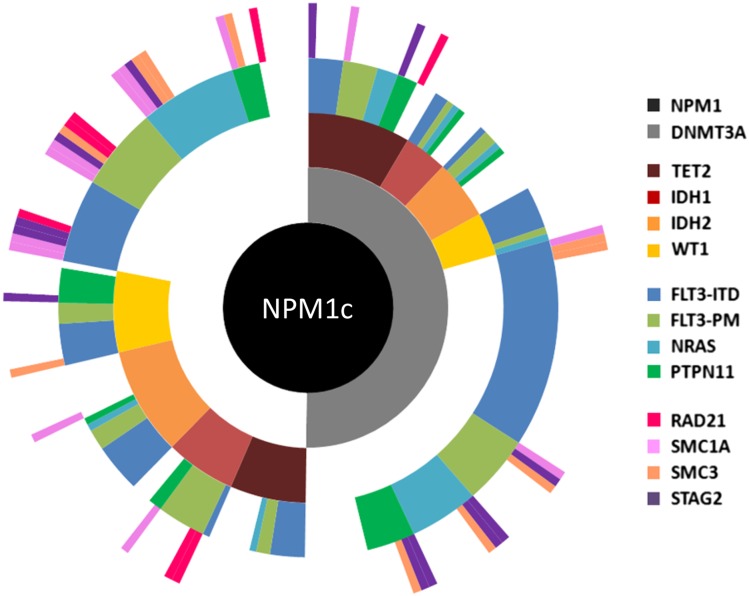
Recent major advances in understanding the molecular basis of acute myeloid leukemia (AML) provide a double-edged sword. Although defining the topology and key features of the molecular landscape are fundamental to development of novel treatment approaches and provide opportunities for greater individualization of therapy, confirmation of the genetic complexity presents a huge challenge to successful translation into routine clinical practice. It is now clear that many genes are recurrently mutated in AML; moreover, individual leukemias harbor multiple mutations and are potentially composed of subclones with differing mutational composition, rendering each patient's AML genetically unique. In order to make sense of the overwhelming mutational data and capitalize on this clinically, it is important to identify (1) critical AML-defining molecular abnormalities that distinguish biological disease entities; (2) mutations, typically arising in subclones, that may influence prognosis but are unlikely to be ideal therapeutic targets; (3) mutations associated with preleukemic clones; and (4) mutations that have been robustly shown to confer independent prognostic information or are therapeutically relevant. The reward of identifying AML-defining molecular lesions present in all leukemic populations (including subclones) has been exemplified by acute promyelocytic leukemia, where successful targeting of the underlying PML-RARα oncoprotein has eliminated the need for chemotherapy for disease cure. Despite the molecular heterogeneity and recognizing that treatment options for other forms of AML are limited, this review will consider the scope for using novel molecular information to improve diagnosis, identify subsets of patients eligible for targeted therapies, refine outcome prediction, and track treatment response.
© 2016 by The American Society of Hematology.
Figures

References
-
- Rowley JD. Identification of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet. 1973;16(2):109–112. - PubMed
-
- Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 1977;309(8010):549–550. - PubMed
-
- Grimwade D, Mrózek K. Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematol Oncol Clin North Am. 2011;25(6):1135–1161, vii. - PubMed
-
- Grimwade D, Hills RK, Moorman AV, et al. National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–365. - PubMed
-
- Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
