

Huntingtin Subcellular Localisation Is Regulated by Kinase Signalling Activity in the StHdhQ111 Model of HD
- PMID: 26660732
- PMCID: PMC4679340
- DOI: 10.1371/journal.pone.0144864
Huntingtin Subcellular Localisation Is Regulated by Kinase Signalling Activity in the StHdhQ111 Model of HD
Erratum in
-
Correction: Huntingtin Subcellular Localisation Is Regulated by Kinase Signalling Activity in the StHdhQ111 Model of HD.PLoS One. 2016 Oct 13;11(10):e0165069. doi: 10.1371/journal.pone.0165069. eCollection 2016. PLoS One. 2016. PMID: 27736977 Free PMC article.
Abstract
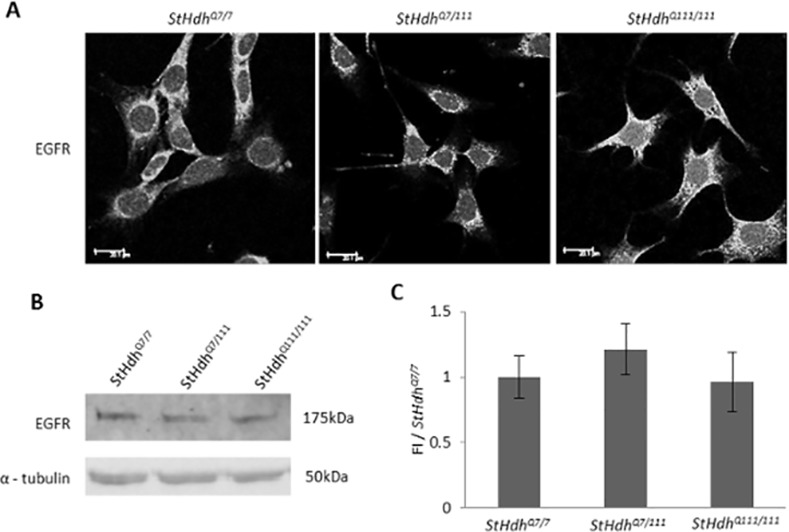
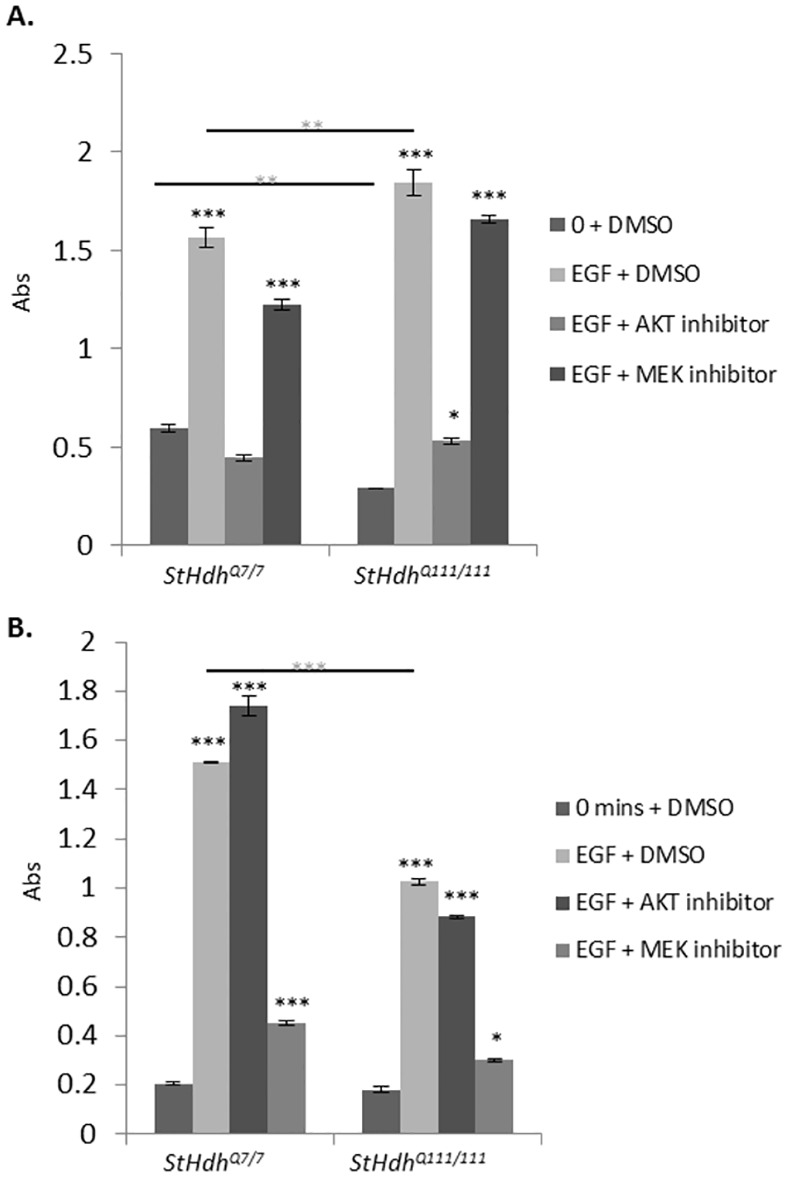
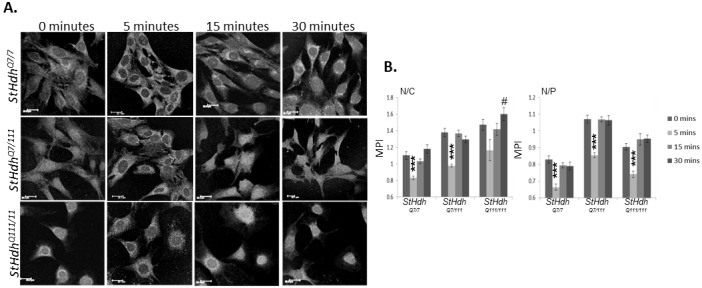
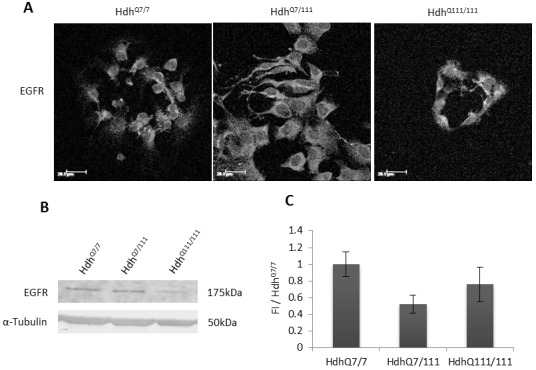
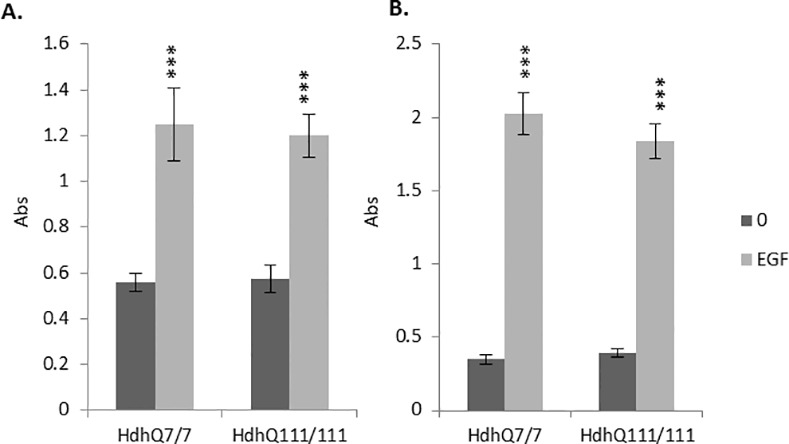
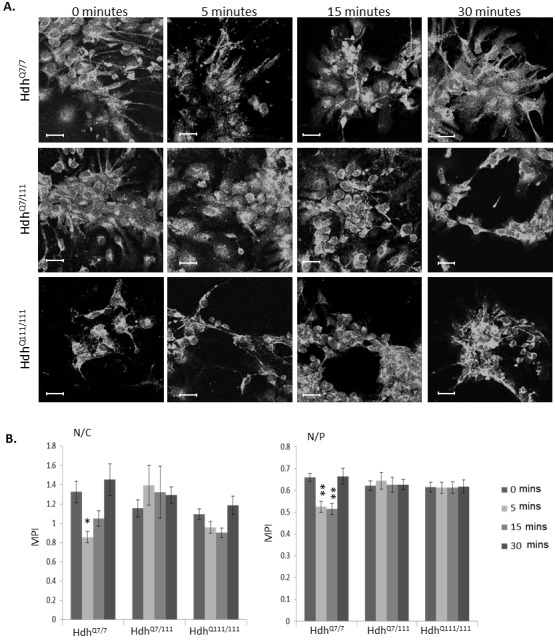
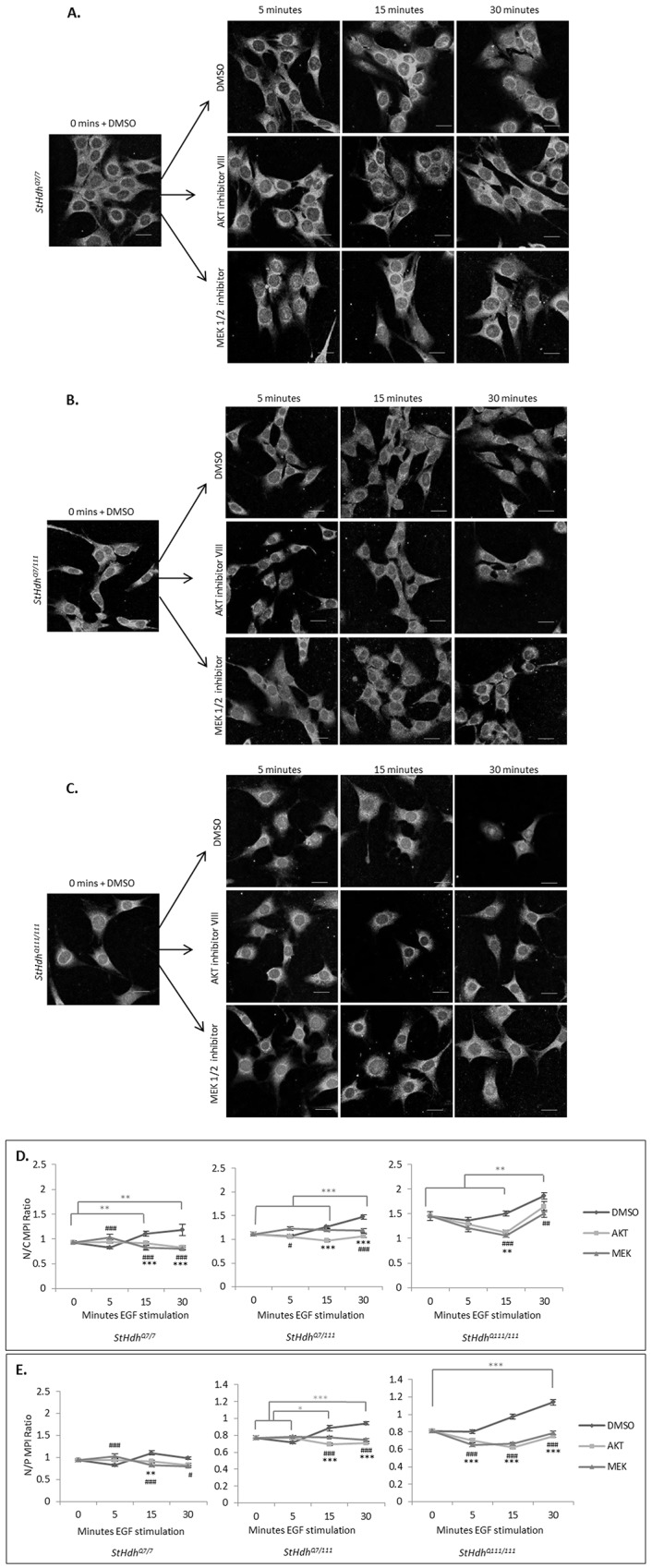
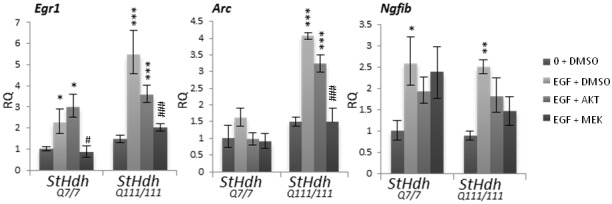
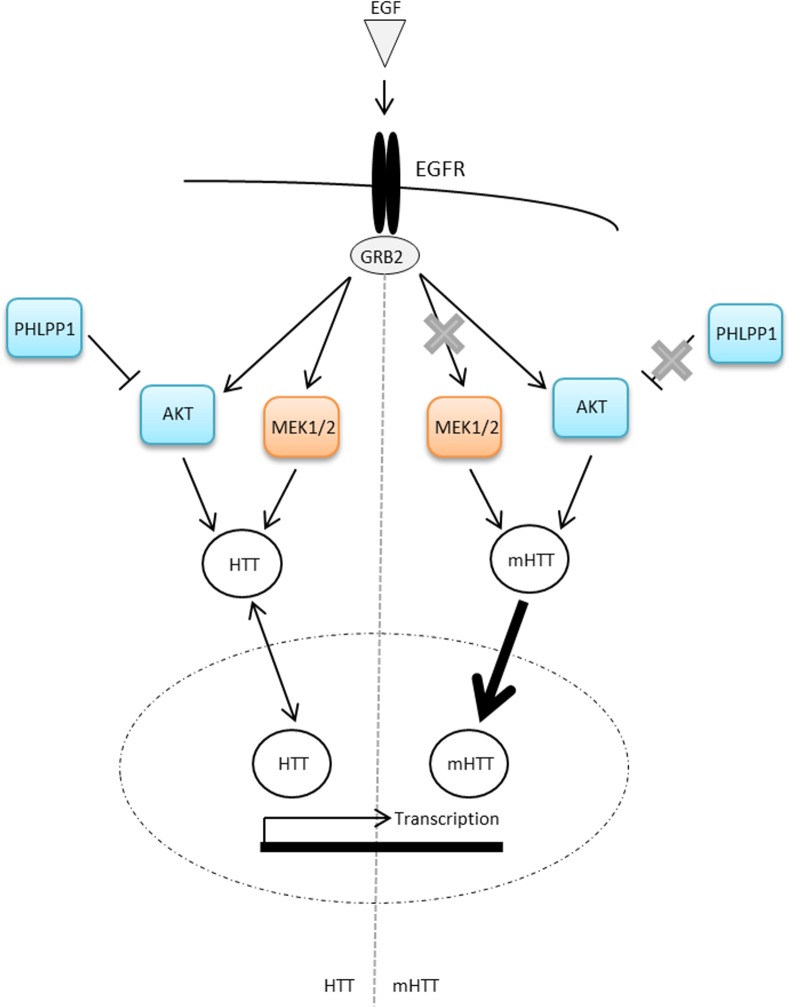
Huntington's disease is a neurodegenerative disorder characterised primarily by motor abnormalities, and is caused by an expanded polyglutamine repeat in the huntingtin protein. Huntingtin dynamically shuttles between subcellular compartments, and the mutant huntingtin protein is mislocalised to cell nuclei, where it may interfere with nuclear functions, such as transcription. However, the mechanism by which mislocalisation of mutant huntingtin occurs is currently unknown. An immortalised embryonic striatal cell model of HD (StHdhQ111) was stimulated with epidermal growth factor in order to determine whether the subcellular localisation of huntingtin is dependent on kinase signalling pathway activation. Aberrant phosphorylation of AKT and MEK signalling pathways was identified in cells carrying mutant huntingtin. Activity within these pathways was found to contribute to the regulation of huntingtin and mutant huntingtin localisation, as well as to the expression of immediate-early genes. We propose that altered kinase signalling is a phenotype of Huntington's disease that occurs prior to cell death; specifically, that altered kinase signalling may influence huntingtin localisation, which in turn may impact upon nuclear processes such as transcriptional regulation. Aiming to restore the balance of activity between kinase signalling networks may therefore prove to be an effective approach to delaying Huntington's disease symptom development and progression.
Conflict of interest statement
Figures
References
-
- Vonsattel JPG, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57: 369–384. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
