

ADAM Proteases and Gastrointestinal Function
- PMID: 26667078
- PMCID: PMC4927194
- DOI: 10.1146/annurev-physiol-021014-071720
ADAM Proteases and Gastrointestinal Function
Abstract
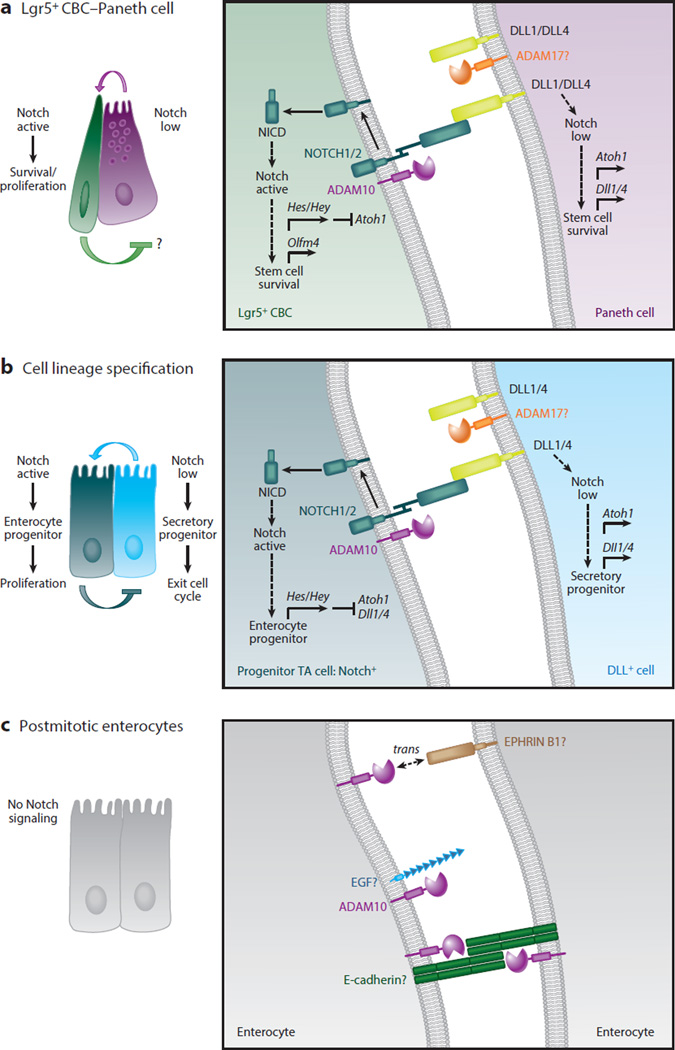
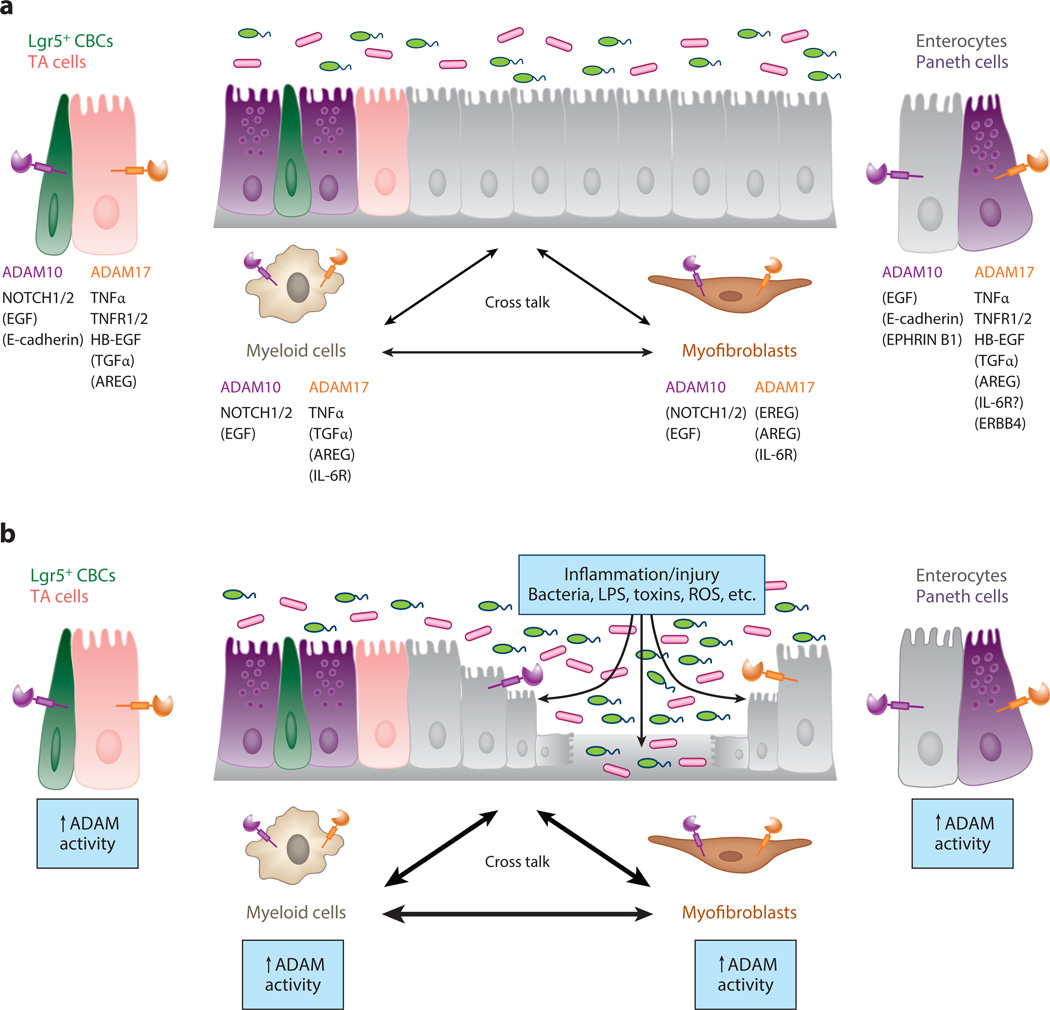
A disintegrin and metalloproteinases (ADAMs) are a family of cell surface proteases that regulate diverse cellular functions, including cell adhesion, migration, cellular signaling, and proteolysis. Proteolytically active ADAMs are responsible for ectodomain shedding of membrane-associated proteins. ADAMs rapidly modulate key cell signaling pathways in response to changes in the extracellular environment (e.g., inflammation) and play a central role in coordinating intercellular communication within the local microenvironment. ADAM10 and ADAM17 are the most studied members of the ADAM family in the gastrointestinal tract. ADAMs regulate many cellular processes associated with intestinal development, cell fate specification, and the maintenance of intestinal stem cell/progenitor populations. Several signaling pathway molecules that undergo ectodomain shedding by ADAMs [e.g., ligands and receptors from epidermal growth factor receptor (EGFR)/ErbB and tumor necrosis factor α (TNFα) receptor (TNFR) families] help drive and control intestinal inflammation and injury/repair responses. Dysregulation of these processes through aberrant ADAM expression or sustained ADAM activity is linked to chronic inflammation, inflammation-associated cancer, and tumorigenesis.
Keywords: ADAM10; ADAM17; EGFR; Notch; TNFα; intestinal stem cells.
Figures
References
-
- Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139:3693–3709. - PubMed
-
- Endres K, Fahrenholz F. Regulation of α-secretase ADAM10 expression and activity. Exp. Brain Res. 2012;217:343–352. - PubMed
-
- Reiss K, Saftig P. The “A disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin. Cell Dev. Biol. 2009;20:126–137. - PubMed
-
- Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011;32:380–387. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
