

Eradication of Large Solid Tumors by Gene Therapy with a T-Cell Receptor Targeting a Single Cancer-Specific Point Mutation
- PMID: 26667491
- PMCID: PMC4891260
- DOI: 10.1158/1078-0432.CCR-15-2361
Eradication of Large Solid Tumors by Gene Therapy with a T-Cell Receptor Targeting a Single Cancer-Specific Point Mutation
Abstract
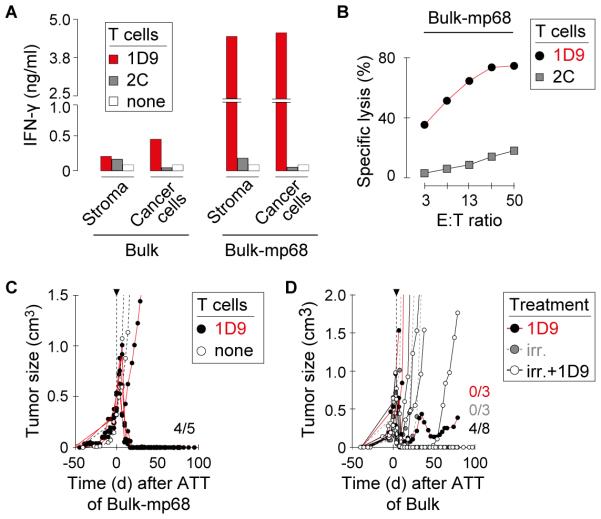
Purpose: Cancers usually contain multiple unique tumor-specific antigens produced by single amino acid substitutions (AAS) and encoded by somatic nonsynonymous single nucleotide substitutions. We determined whether adoptively transferred T cells can reject large, well-established solid tumors when engineered to express a single type of T-cell receptor (TCR) that is specific for a single AAS.
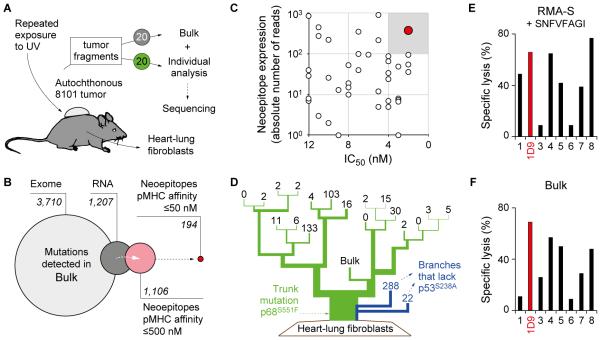
Experimental design: By exome and RNA sequencing of an UV-induced tumor, we identified an AAS in p68 (mp68), a co-activator of p53. This AAS seemed to be an ideal tumor-specific neoepitope because it is encoded by a trunk mutation in the primary autochthonous cancer and binds with highest affinity to the MHC. A high-avidity mp68-specific TCR was used to genetically engineer T cells as well as to generate TCR-transgenic mice for adoptive therapy.
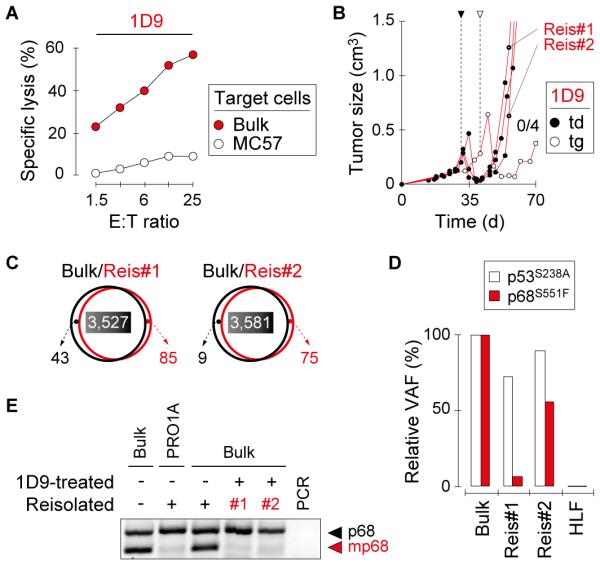
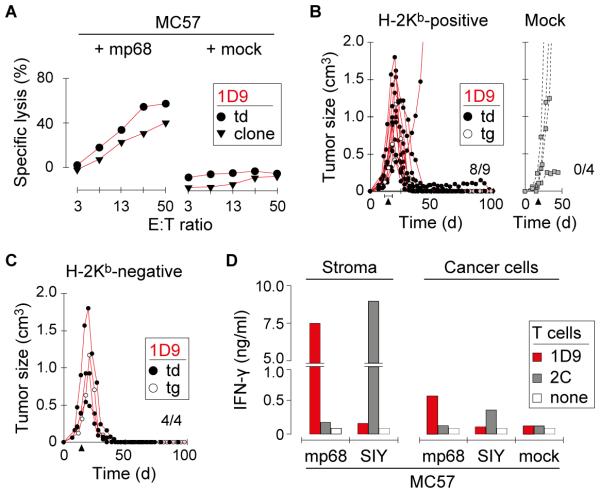
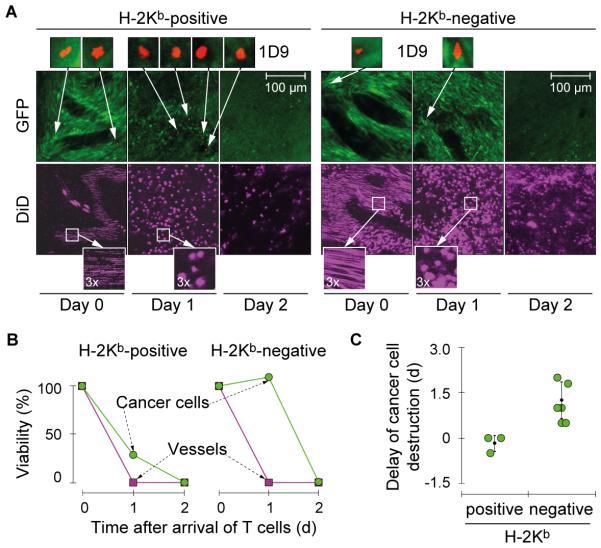
Results: When the neoepitope was expressed at high levels and by all cancer cells, their direct recognition sufficed to destroy intratumor vessels and eradicate large, long-established solid tumors. When the neoepitope was targeted as autochthonous antigen, T cells caused cancer regression followed by escape of antigen-negative variants. Escape could be thwarted by expressing the antigen at increased levels in all cancer cells or by combining T-cell therapy with local irradiation. Therapeutic efficacies of TCR-transduced and TCR-transgenic T cells were similar.
Conclusions: Gene therapy with a single TCR targeting a single AAS can eradicate large established cancer, but a uniform expression and/or sufficient levels of the targeted neoepitope or additional therapy are required to overcome tumor escape. Clin Cancer Res; 22(11); 2734-43. ©2015 AACRSee related commentary by Liu, p. 2602.
©2015 American Association for Cancer Research.
Figures
Comment in
-
Neoantigen: A Long March toward Cancer Immunotherapy.Clin Cancer Res. 2016 Jun 1;22(11):2602-4. doi: 10.1158/1078-0432.CCR-15-3170. Epub 2016 Mar 22. Clin Cancer Res. 2016. PMID: 27006495 Free PMC article.
References
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. - PubMed
-
- Monach PA, Meredith SC, Siegel CT, Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2:45–59. - PubMed
-
- Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
