

Common errors in mass spectrometry-based analysis of post-translational modifications
- PMID: 26667783
- PMCID: PMC5548100
- DOI: 10.1002/pmic.201500355
Common errors in mass spectrometry-based analysis of post-translational modifications
Abstract
Mass spectrometry (MS) is a powerful tool to analyze complex mixtures of proteins in a high-throughput fashion. Proteome analysis has already become a routine task in biomedical research with the emergence of proteomics core facilities in most research institutions. Post-translational modifications (PTMs) represent a mechanism by which complex biological processes are orchestrated dynamically at the systems level. MS is rapidly becoming popular to discover new modifications and novel sites of known PTMs, revolutionizing the current understanding of diverse signaling pathways and biological processes. However, MS-based analysis of PTMs has its own caveats and pitfalls that can lead to erroneous conclusions. Here, we review the most common errors in MS-based PTM analyses with the goal of adopting strategies that maximize correct interpretation in the context of biological questions that are being addressed. Finally, we provide suggestions that should help mass spectrometrists, bioinformaticians and biologists to perform and interpret MS-based PTM analyses more accurately.
Keywords: Immonium; Localization; PTM; SUMO; Signature; Technology; Ubiquitin.
© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Conflict of interest statement
The authors have declared no conflict of interest.
Figures
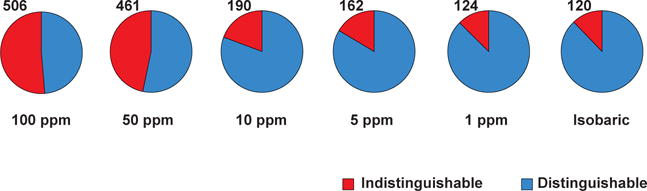
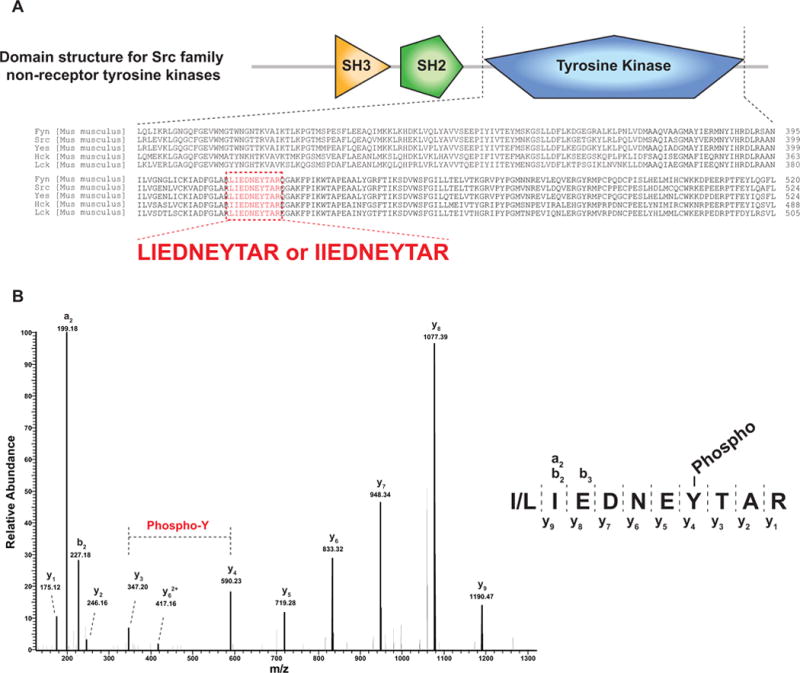
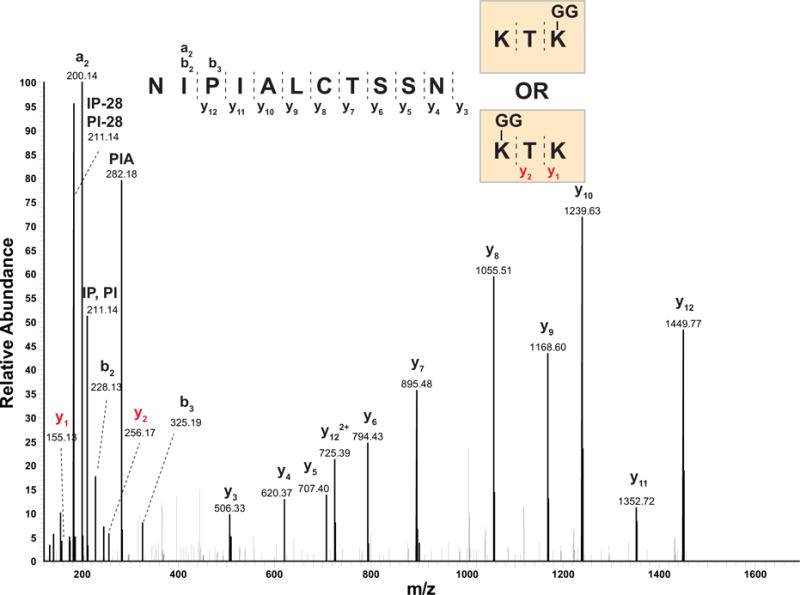
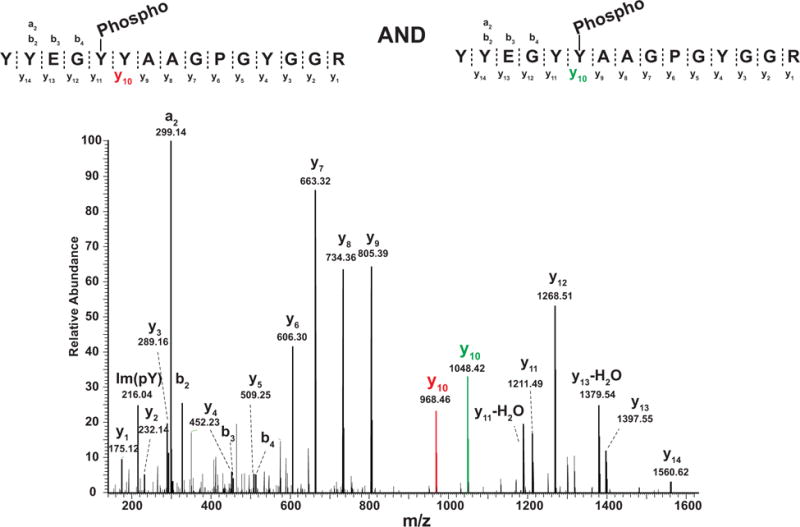
Similar articles
-
Prediction of novel modifications by unrestrictive search of tandem mass spectra.J Proteome Res. 2009 Oct;8(10):4418-27. doi: 10.1021/pr9001146. J Proteome Res. 2009. PMID: 19658439
-
Non-parametric Bayesian approach to post-translational modification refinement of predictions from tandem mass spectrometry.Bioinformatics. 2013 Apr 1;29(7):821-9. doi: 10.1093/bioinformatics/btt056. Epub 2013 Feb 17. Bioinformatics. 2013. PMID: 23419374
-
Identification, Quantification, and Site Localization of Protein Posttranslational Modifications via Mass Spectrometry-Based Proteomics.Adv Exp Med Biol. 2016;919:345-382. doi: 10.1007/978-3-319-41448-5_17. Adv Exp Med Biol. 2016. PMID: 27975226 Review.
-
Identification of ubiquitin/ubiquitin-like protein modification from tandem mass spectra with various PTMs.BMC Bioinformatics. 2011 Dec 14;12 Suppl 14(Suppl 14):S8. doi: 10.1186/1471-2105-12-S14-S8. BMC Bioinformatics. 2011. PMID: 22373085 Free PMC article.
-
From proteome lists to biological impact--tools and strategies for the analysis of large MS data sets.Proteomics. 2010 Mar;10(6):1270-83. doi: 10.1002/pmic.200900365. Proteomics. 2010. PMID: 20077408 Review.
Cited by
-
Origins, Technological Advancement, and Applications of Peptidomics.Methods Mol Biol. 2024;2758:3-47. doi: 10.1007/978-1-0716-3646-6_1. Methods Mol Biol. 2024. PMID: 38549006
-
Dynamic acylome reveals metabolite driven modifications in Syntrophomonas wolfei.Front Microbiol. 2022 Nov 7;13:1018220. doi: 10.3389/fmicb.2022.1018220. eCollection 2022. Front Microbiol. 2022. PMID: 36419437 Free PMC article.
-
Leveraging Immonium Ions for Targeting Acyl-Lysine Modifications in Proteomic Datasets.Proteomics. 2021 Feb;21(3-4):e2000111. doi: 10.1002/pmic.202000111. Epub 2020 Sep 25. Proteomics. 2021. PMID: 32896103 Free PMC article.
-
SP3-Enabled Rapid and High Coverage Chemoproteomic Identification of Cell-State-Dependent Redox-Sensitive Cysteines.Mol Cell Proteomics. 2022 Apr;21(4):100218. doi: 10.1016/j.mcpro.2022.100218. Epub 2022 Feb 25. Mol Cell Proteomics. 2022. PMID: 35219905 Free PMC article.
-
Beyond mass spectrometry, the next step in proteomics.Sci Adv. 2020 Jan 10;6(2):eaax8978. doi: 10.1126/sciadv.aax8978. eCollection 2020 Jan. Sci Adv. 2020. PMID: 31950079 Free PMC article. Review.
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Cravatt BF, Simon GM, Yates JR., Iii The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. - PubMed
-
- Montecchi-Palazzi L, Beavis R, Binz PA, Chalkley RJ, et al. The PSI-MOD community standard for representation of protein modification data. Nat Biotechnol. 2008;26:864–866. - PubMed
-
- Garavelli JS. The RESID Database of Protein Modifications as a resource and annotation tool. Proteomics. 2004;4:1527–1533. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
