

Transcriptome-wide high-throughput deep m(6)A-seq reveals unique differential m(6)A methylation patterns between three organs in Arabidopsis thaliana
- PMID: 26667818
- PMCID: PMC4714525
- DOI: 10.1186/s13059-015-0839-2
Transcriptome-wide high-throughput deep m(6)A-seq reveals unique differential m(6)A methylation patterns between three organs in Arabidopsis thaliana
Abstract
Background: m(6)A is a ubiquitous RNA modification in eukaryotes. Transcriptome-wide m(6)A patterns in Arabidopsis have been assayed recently. However, differential m(6)A patterns between organs have not been well characterized.
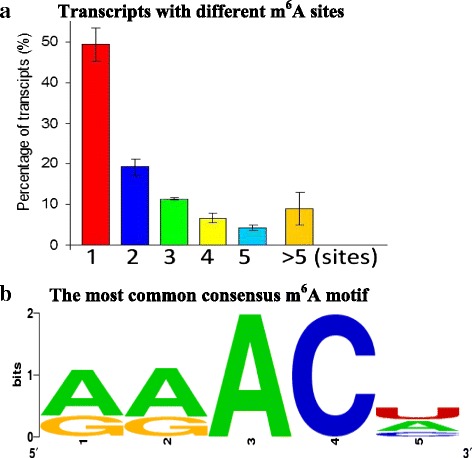
Results: Over two-third of the transcripts in Arabidopsis are modified by m(6)A. In contrast to a recent observation of m(6)A enrichment in 5' mRNA, we find that m(6)A is distributed predominantly near stop codons. Interestingly, 85 % of the modified transcripts show high m(6)A methylation extent compared to their transcript level. The 290 highly methylated transcripts are mainly associated with transporters, stress responses, redox, regulation factors, and some non-coding RNAs. On average, the proportion of transcripts showing differential methylation between two plant organs is higher than that showing differential transcript levels. The transcripts with extensively higher m(6)A methylation in an organ are associated with the unique biological processes of this organ, suggesting that m(6)A may be another important contributor to organ differentiation in Arabidopsis. Highly expressed genes are relatively less methylated and vice versa, and different RNAs have distinct m(6)A patterns, which hint at mRNA fate. Intriguingly, most of the transposable element transcripts maintained a fragmented form with a relatively low transcript level and high m(6)A methylation in the cells.
Conclusions: This is the first study to comprehensively analyze m(6)A patterns in a variety of RNAs, the relationship between transcript level and m(6)A methylation extent, and differential m(6)A patterns across organs in Arabidopsis.
Figures









References
-
- Grosjean H. Fine-tuning of RNA functions by modification and editing. Berlin: Springer-Verlag; 2005.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
