

Desmoplakin Variants Are Associated with Idiopathic Pulmonary Fibrosis
- PMID: 26669357
- PMCID: PMC4872666
- DOI: 10.1164/rccm.201509-1863OC
Desmoplakin Variants Are Associated with Idiopathic Pulmonary Fibrosis
Abstract
Rationale: Sequence variation, methylation differences, and transcriptional changes in desmoplakin (DSP) have been observed in patients with idiopathic pulmonary fibrosis (IPF).
Objectives: To identify novel variants in DSP associated with IPF and to characterize the relationship of these IPF sequence variants with DSP gene expression in human lung.
Methods: A chromosome 6 locus (7,370,061-7,606,946) was sequenced in 230 subjects with IPF and 228 control subjects. Validation genotyping of disease-associated variants was conducted in 936 subjects with IPF and 936 control subjects. DSP gene expression was measured in lung tissue from 334 subjects with IPF and 201 control subjects.
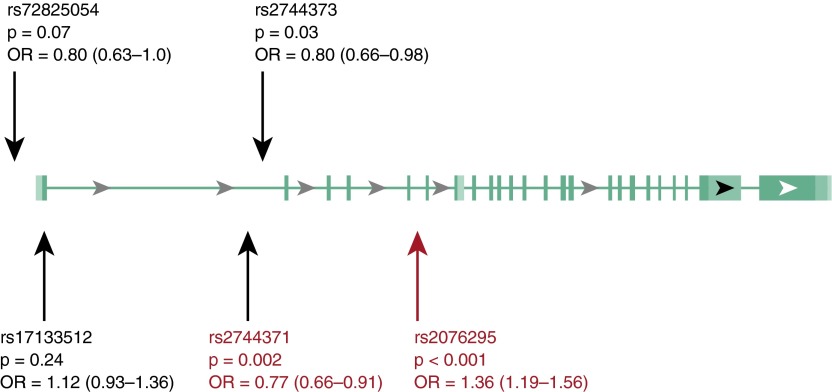
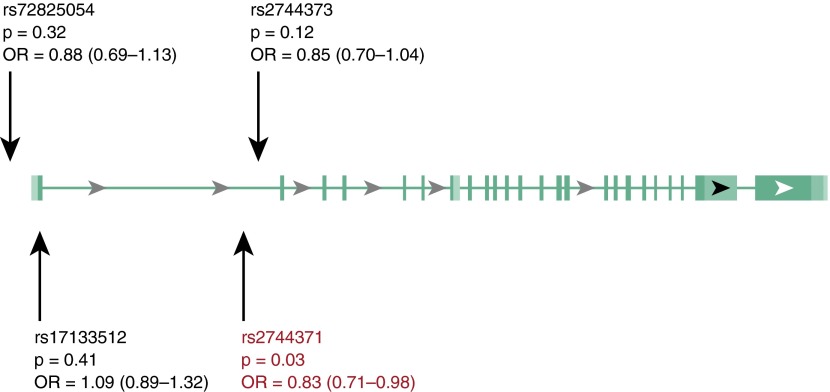
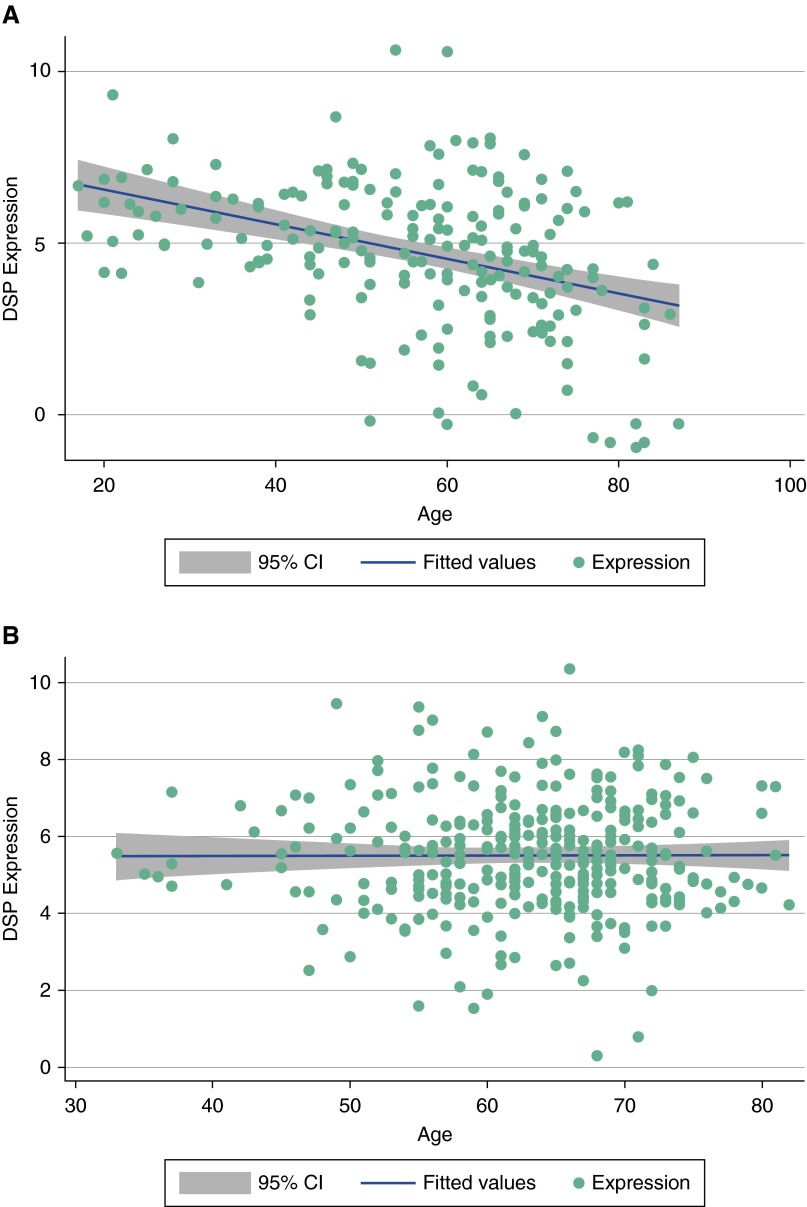
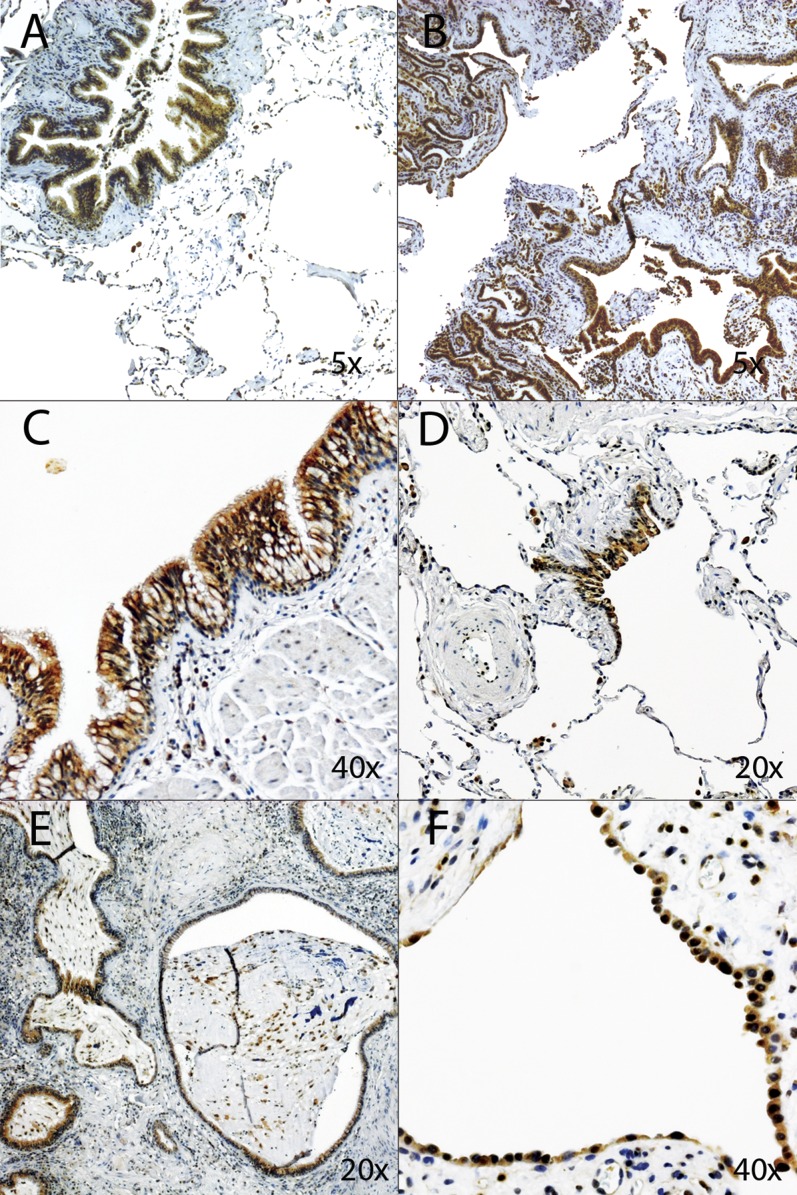
Measurements and main results: We identified 23 sequence variants in the chromosome 6 locus associated with IPF. Genotyping of selected variants in our validation cohort revealed that noncoding intron 1 variant rs2744371 (odds ratio = 0.77, 95% confidence interval [CI] = 0.66-0.91, P = 0.002) is protective for IPF, and a previously described IPF-associated intron 5 variant (rs2076295) is associated with increased risk of IPF (odds ratio = 1.36, 95% CI = 1.19-1.56, P < 0.001) after controlling for sex and age. DSP expression is 2.3-fold increased (95% CI = 1.91-2.71) in IPF lung tissue (P < 0.0001). Only the minor allele at rs2076295 is associated with decreased DSP expression (P = 0.001). Staining of fibrotic and normal human lung tissue localized DSP to airway epithelia.
Conclusions: Sequence variants in DSP are associated with IPF, and rs2076295 genotype is associated with differential expression of DSP in the lung. DSP expression is increased in IPF lung and concentrated in the airway epithelia, suggesting a potential role for DSP in the pathogenesis of IPF.
Keywords: desmoplakin; genetics; idiopathic interstitial pneumonia; idiopathic pulmonary fibrosis; intron.
Figures


Comment in
-
The Airway in Idiopathic Pulmonary Fibrosis: Protecting the Lung or Promoting Disease?Am J Respir Crit Care Med. 2016 May 15;193(10):1081-2. doi: 10.1164/rccm.201601-0055ED. Am J Respir Crit Care Med. 2016. PMID: 27174477 Free PMC article. No abstract available.
References
-
- King TE, Jr, Albera C, Bradford WZ, Costabel U, du Bois RM, Leff JA, Nathan SD, Sahn SA, Valeyre D, Noble PW. All-cause mortality rate in patients with idiopathic pulmonary fibrosis: implications for the design and execution of clinical trials. Am J Respir Crit Care Med. 2014;189:825–831. - PubMed
-
- King TE, Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001;164:1171–1181. - PubMed
-
- Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277–284. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
