

Common Viral Integration Sites Identified in Avian Leukosis Virus-Induced B-Cell Lymphomas
- PMID: 26670384
- PMCID: PMC4701831
- DOI: 10.1128/mBio.01863-15
Common Viral Integration Sites Identified in Avian Leukosis Virus-Induced B-Cell Lymphomas
Abstract
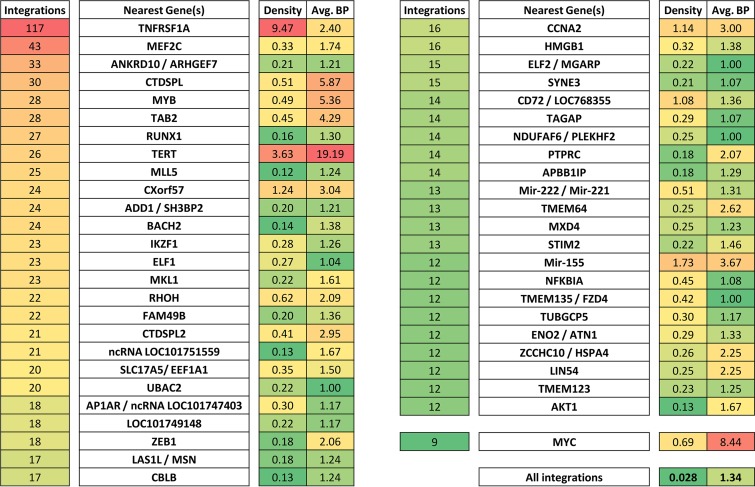
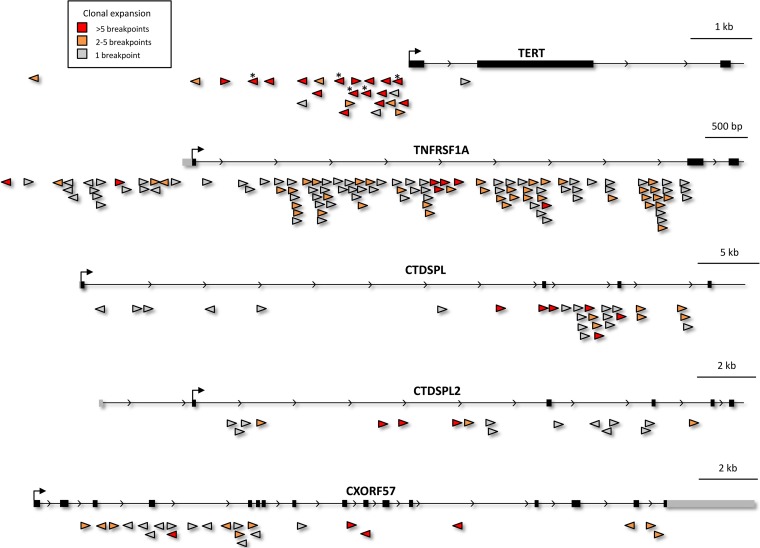
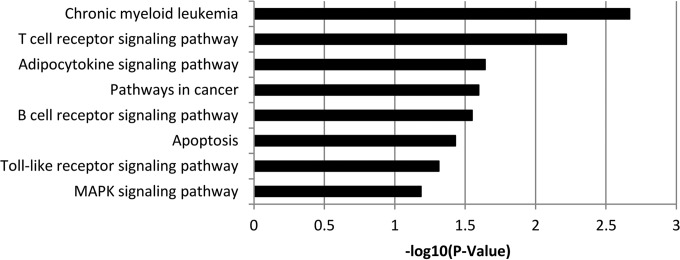
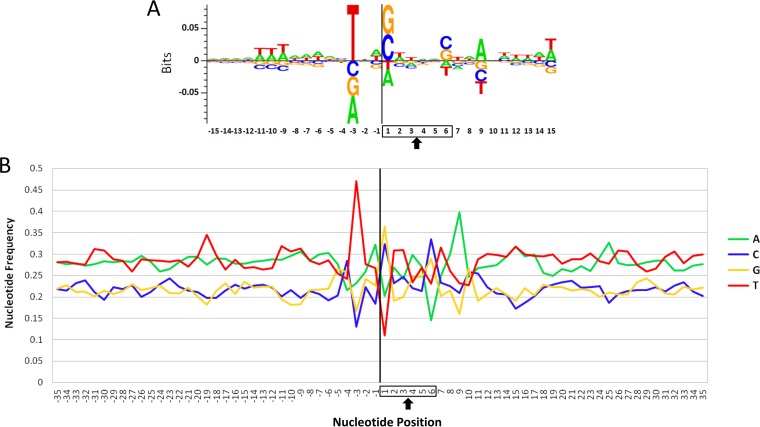
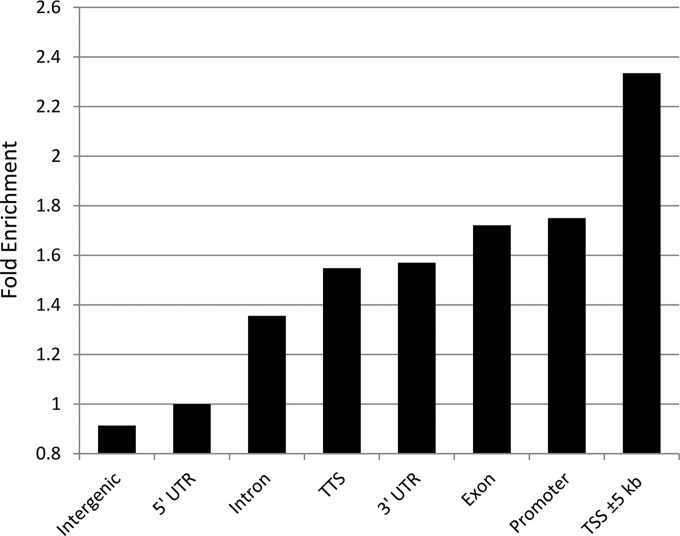
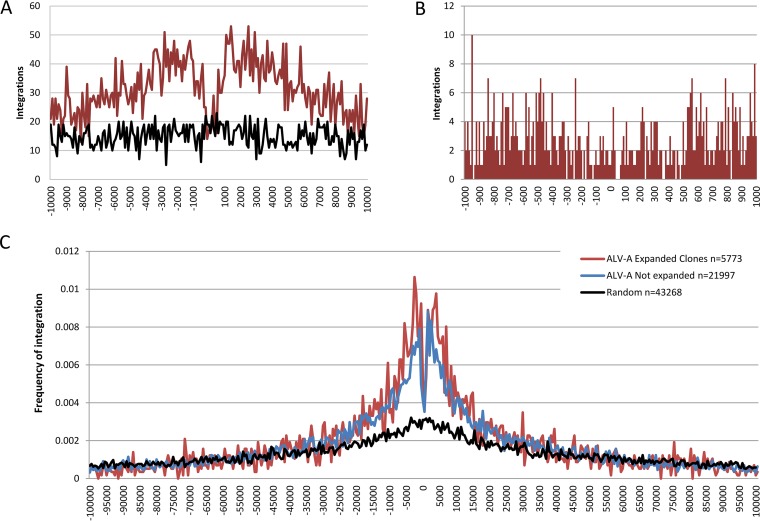
Avian leukosis virus (ALV) induces B-cell lymphoma and other neoplasms in chickens by integrating within or near cancer genes and perturbing their expression. Four genes--MYC, MYB, Mir-155, and TERT--have previously been identified as common integration sites in these virus-induced lymphomas and are thought to play a causal role in tumorigenesis. In this study, we employ high-throughput sequencing to identify additional genes driving tumorigenesis in ALV-induced B-cell lymphomas. In addition to the four genes implicated previously, we identify other genes as common integration sites, including TNFRSF1A, MEF2C, CTDSPL, TAB2, RUNX1, MLL5, CXorf57, and BACH2. We also analyze the genome-wide ALV integration landscape in vivo and find increased frequency of ALV integration near transcriptional start sites and within transcripts. Previous work has shown ALV prefers a weak consensus sequence for integration in cultured human cells. We confirm this consensus sequence for ALV integration in vivo in the chicken genome.
Importance: Avian leukosis virus induces B-cell lymphomas in chickens. Earlier studies showed that ALV can induce tumors through insertional mutagenesis, and several genes have been implicated in the development of these tumors. In this study, we use high-throughput sequencing to reveal the genome-wide ALV integration landscape in ALV-induced B-cell lymphomas. We find elevated levels of ALV integration near transcription start sites and use common integration site analysis to greatly expand the number of genes implicated in the development of these tumors. Interestingly, we identify several genes targeted by viral insertions that have not been previously shown to be involved in cancer.
Copyright © 2015 Justice et al.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
