

Refining the continuum of CFTR-associated disorders in the era of newborn screening
- PMID: 26671754
- PMCID: PMC5389888
- DOI: 10.1111/cge.12711
Refining the continuum of CFTR-associated disorders in the era of newborn screening
Abstract
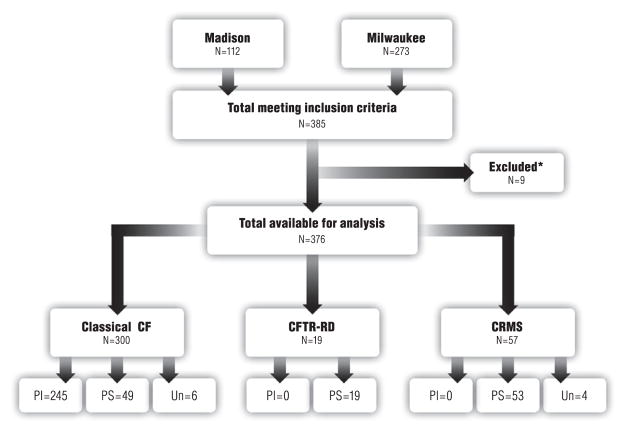
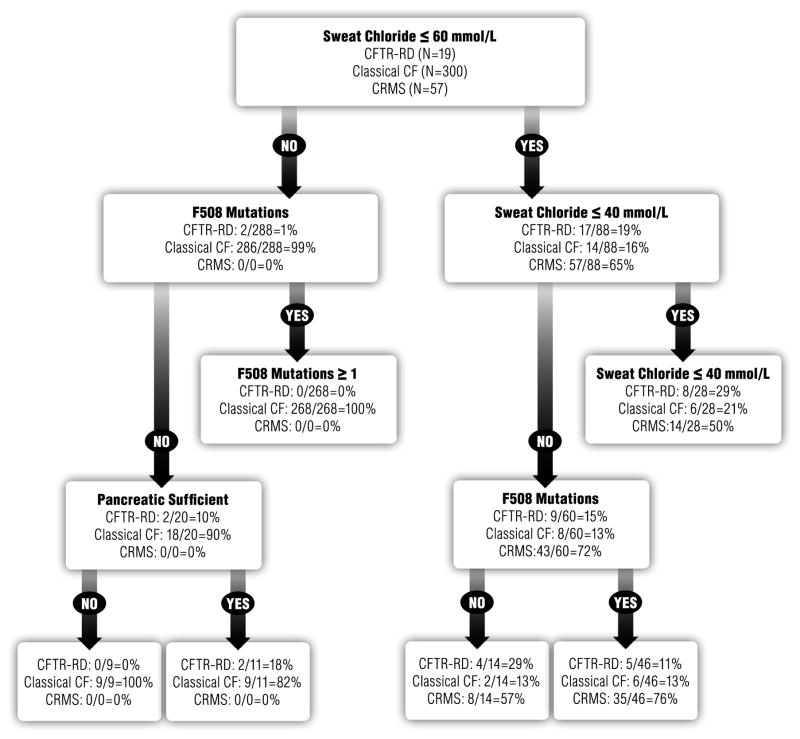
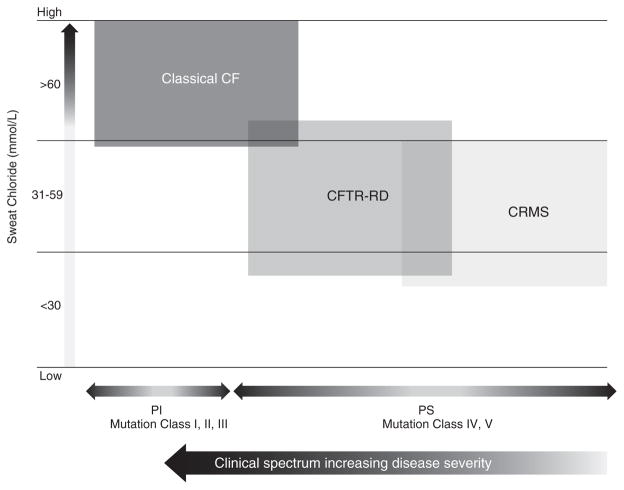
Clinical heterogeneity in cystic fibrosis (CF) often causes diagnostic uncertainty in infants without symptoms and in older patients with milder phenotypes. We performed a cross-sectional evaluation of a comprehensive set of clinical and laboratory descriptors in a physician-defined cohort (N = 376; Children's Hospital of Wisconsin and the American Family Children's Hospital CF centers in Milwaukee and Madison, WI, USA) to determine the robustness of categorizing CF (N = 300), cystic fibrosis transmembrane conductance regulator (CFTR)-related disorder (N = 19), and CFTR-related (CRMS) metabolic syndrome (N = 57) according to current consensus guidelines. Outcome measures included patient demographics, clinical measures, sweat chloride levels, CFTR genotype, age at diagnosis, airway microbiology, pancreatic function, infection, and nutritional status. The CF cohort had a significantly higher median sweat chloride level (105 mmol/l) than CFTR-related disorder patients (43 mmol/l) and CFTR-related metabolic syndrome patients (35 mmol/l; p ≤ 0.001). Patient groups significantly differed in pancreatic sufficiency, immunoreactive trypsinogen levels, sweat chloride values, genotype, and positive Pseudomonas aeruginosa cultures (p ≤ 0.001). An automated classification algorithm using recursive partitioning demonstrated concordance between physician diagnoses and consensus guidelines. Our analysis suggests that integrating clinical information with sweat chloride levels, CFTR genotype, and pancreatic sufficiency provides a context for continued longitudinal monitoring of patients for personalized and effective treatment.
Keywords: CFTR phenotype-genotype correlation; CFTR-opathies; cystic fibrosis; cystic fibrosis metabolic syndrome; cystic fibrosis-related disorder.
© 2015 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ooi CY, Castellani C, Keenan K, et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics. 2015;135:e1377–e1385. - PubMed
-
- Groman JD, Meyer ME, Wilmott RW, Zeitlin PL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. New Engl J Med. 2002;347:401–407. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
