

Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations
- PMID: 26675346
- PMCID: PMC4768426
- DOI: 10.1182/blood-2015-10-674572
Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations
Abstract
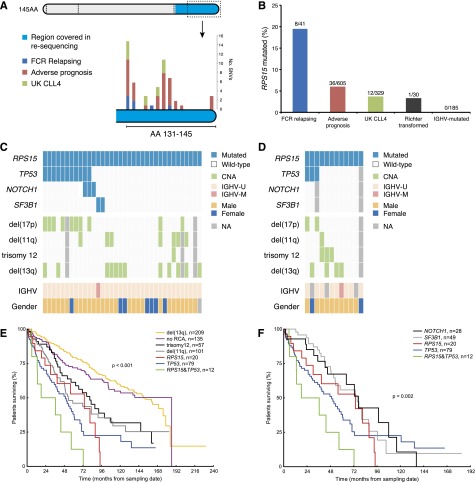
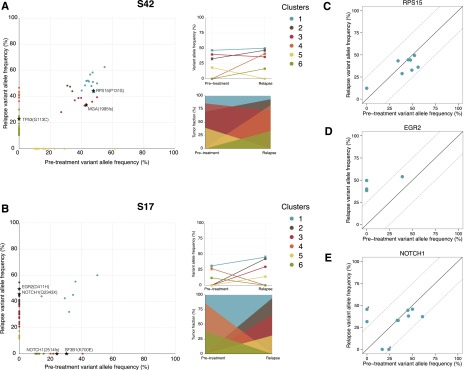
Fludarabine, cyclophosphamide, and rituximab (FCR) is first-line treatment of medically fit chronic lymphocytic leukemia (CLL) patients; however, despite good response rates, many patients eventually relapse. Although recent high-throughput studies have identified novel recurrent genetic lesions in adverse prognostic CLL, the mechanisms leading to relapse after FCR therapy are not completely understood. To gain insight into this issue, we performed whole-exome sequencing of sequential samples from 41 CLL patients who were uniformly treated with FCR but relapsed after a median of 2 years. In addition to mutations with known adverse-prognostic impact (TP53, NOTCH1, ATM, SF3B1, NFKBIE, and BIRC3), a large proportion of cases (19.5%) harbored mutations in RPS15, a gene encoding a component of the 40S ribosomal subunit. Extended screening, totaling 1119 patients, supported a role for RPS15 mutations in aggressive CLL, with one-third of RPS15-mutant cases also carrying TP53 aberrations. In most cases, selection of dominant, relapse-specific subclones was observed over time. However, RPS15 mutations were clonal before treatment and remained stable at relapse. Notably, all RPS15 mutations represented somatic missense variants and resided within a 7 amino-acid, evolutionarily conserved region. We confirmed the recently postulated direct interaction between RPS15 and MDM2/MDMX and transient expression of mutant RPS15 revealed defective regulation of endogenous p53 compared with wild-type RPS15. In summary, we provide novel insights into the heterogeneous genetic landscape of CLL relapsing after FCR treatment and highlight a novel mechanism underlying clinical aggressiveness involving a mutated ribosomal protein, potentially representing an early genetic lesion in CLL pathobiology.
© 2016 by The American Society of Hematology.
Figures


Comment in
-
Ribosomal revelation.Blood. 2016 Feb 25;127(8):958-9. doi: 10.1182/blood-2015-12-688994. Blood. 2016. PMID: 26917735 No abstract available.
References
-
- Hallek M, Fischer K, Fingerle-Rowson G, et al. International Group of Investigators; German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. - PubMed
-
- Wierda W, O’Brien S, Wen S, et al. Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab for relapsed and refractory chronic lymphocytic leukemia. J Clin Oncol. 2005;23(18):4070–4078. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
