

Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy
- PMID: 26676145
- PMCID: PMC4698395
- DOI: 10.1152/physrev.00007.2015
Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy
Abstract
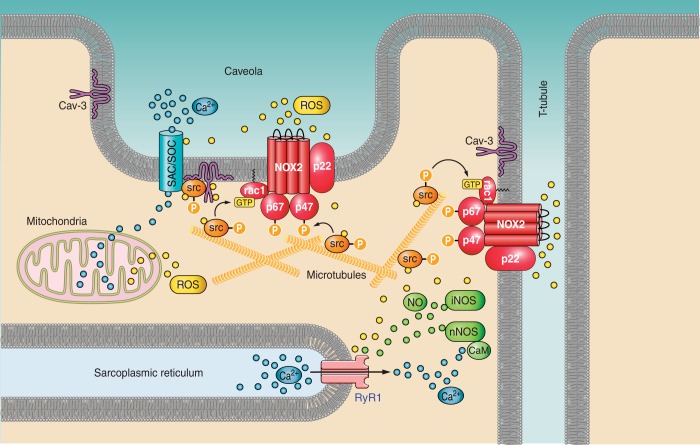
Dystrophin is a long rod-shaped protein that connects the subsarcolemmal cytoskeleton to a complex of proteins in the surface membrane (dystrophin protein complex, DPC), with further connections via laminin to other extracellular matrix proteins. Initially considered a structural complex that protected the sarcolemma from mechanical damage, the DPC is now known to serve as a scaffold for numerous signaling proteins. Absence or reduced expression of dystrophin or many of the DPC components cause the muscular dystrophies, a group of inherited diseases in which repeated bouts of muscle damage lead to atrophy and fibrosis, and eventually muscle degeneration. The normal function of dystrophin is poorly defined. In its absence a complex series of changes occur with multiple muscle proteins showing reduced or increased expression or being modified in various ways. In this review, we will consider the various proteins whose expression and function is changed in muscular dystrophies, focusing on Ca(2+)-permeable channels, nitric oxide synthase, NADPH oxidase, and caveolins. Excessive Ca(2+) entry, increased membrane permeability, disordered caveolar function, and increased levels of reactive oxygen species are early changes in the disease, and the hypotheses for these phenomena will be critically considered. The aim of the review is to define the early damage pathways in muscular dystrophy which might be appropriate targets for therapy designed to minimize the muscle degeneration and slow the progression of the disease.
Copyright © 2016 the American Physiological Society.
Figures






References
-
- Aartsma-Rus A, Muntoni F. 194th ENMC International Workshop. 3rd ENMC workshop on exon skipping: towards clinical application of antisense-mediated exon skipping for Duchenne muscular dystrophy 8–10 December 2012, Naarden, The Netherlands. Neuromuscul Disord 23: 934–944, 2013. - PubMed
-
- Abramovici H, Gee SH. Morphological changes and spatial regulation of diacylglycerol kinase-zeta, syntrophins, and Rac1 during myoblast fusion. Cell Motil Cytoskeleton 64: 549–567, 2007. - PubMed
-
- Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron 11: 531–540, 1993. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous