

Mouse Hepatitis Virus Infection Remodels Connexin43-Mediated Gap Junction Intercellular Communication In Vitro and In Vivo
- PMID: 26676788
- PMCID: PMC4810703
- DOI: 10.1128/JVI.02420-15
Mouse Hepatitis Virus Infection Remodels Connexin43-Mediated Gap Junction Intercellular Communication In Vitro and In Vivo
Abstract
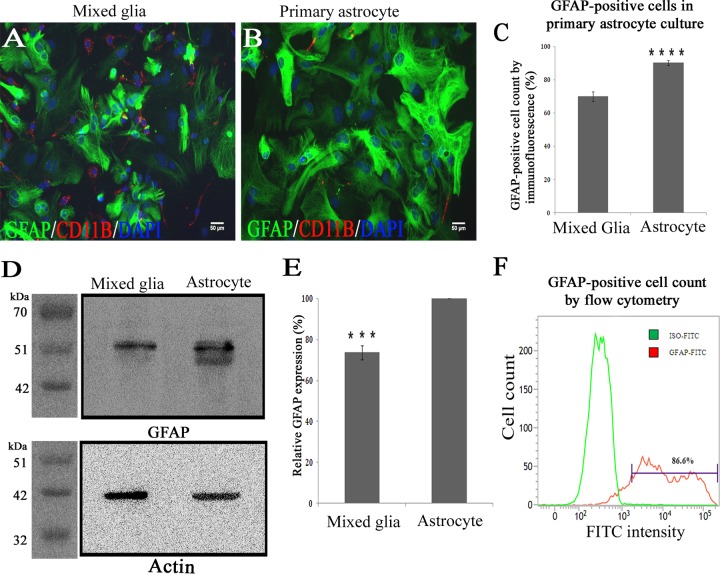
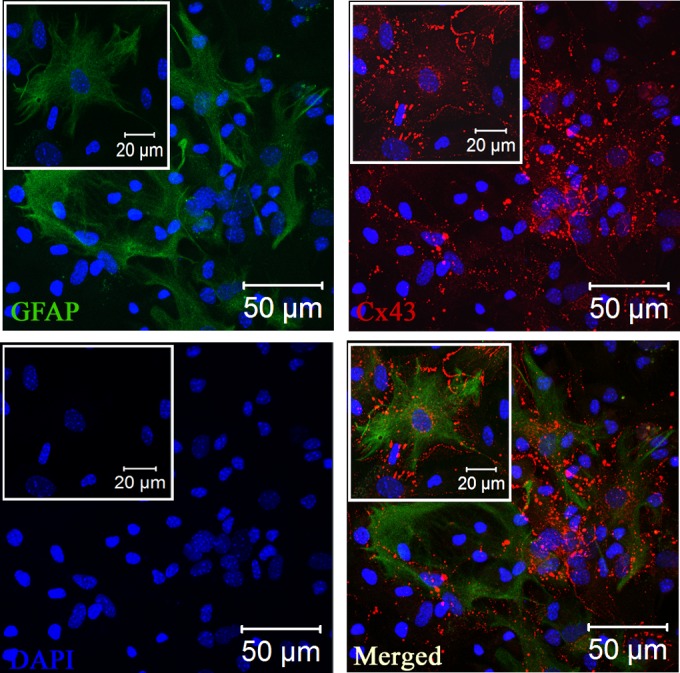
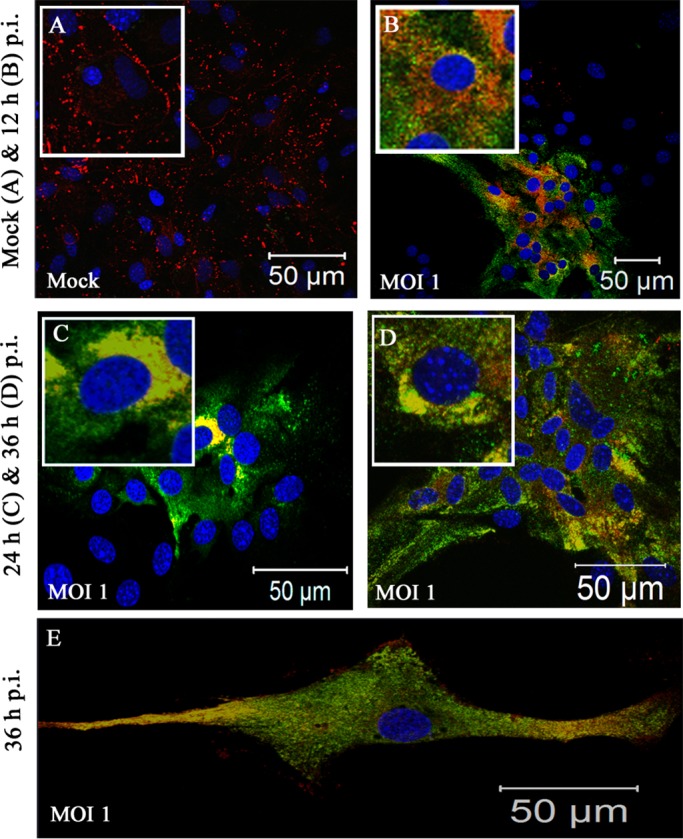
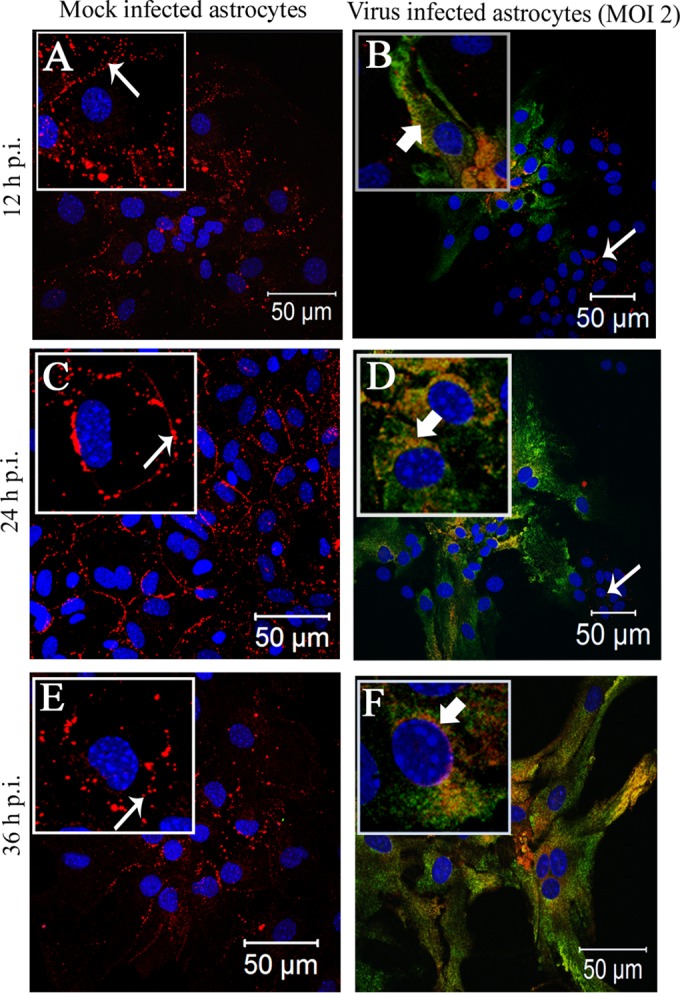
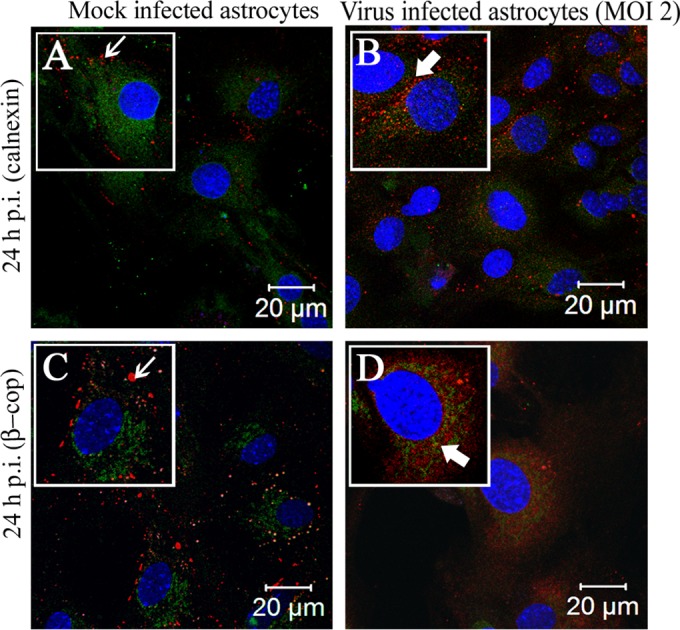
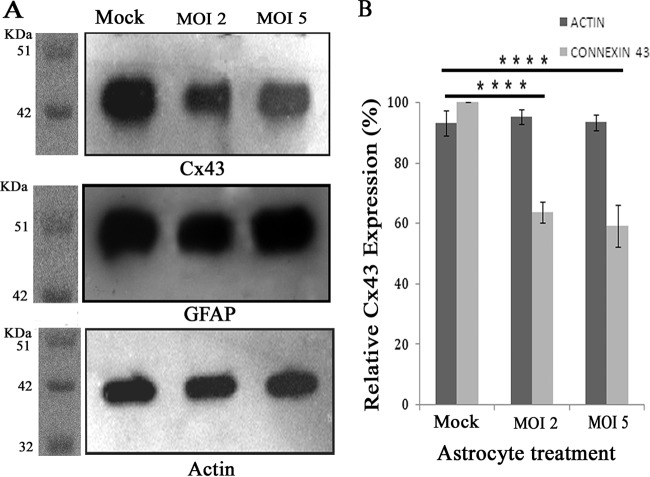
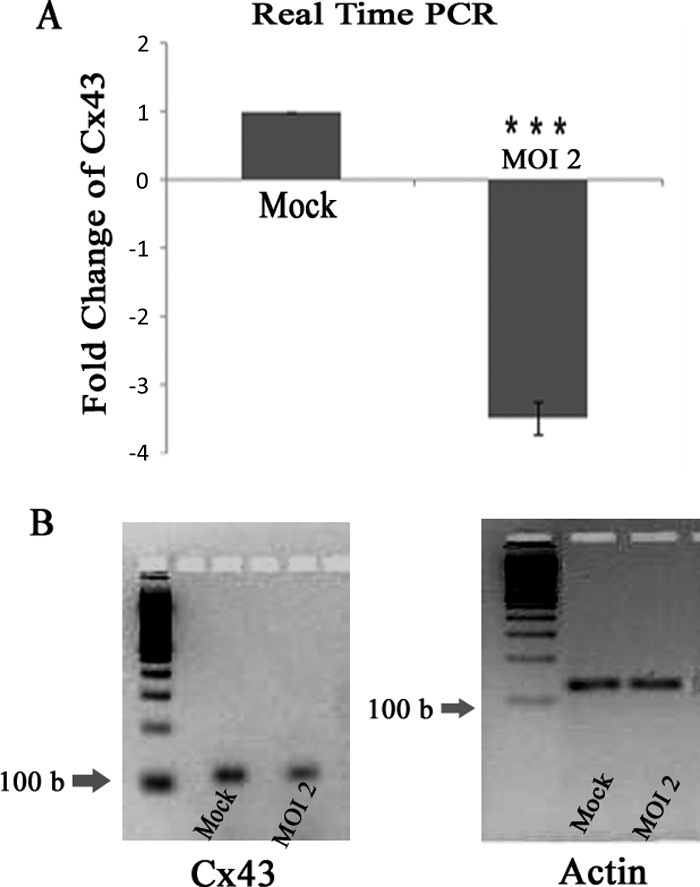
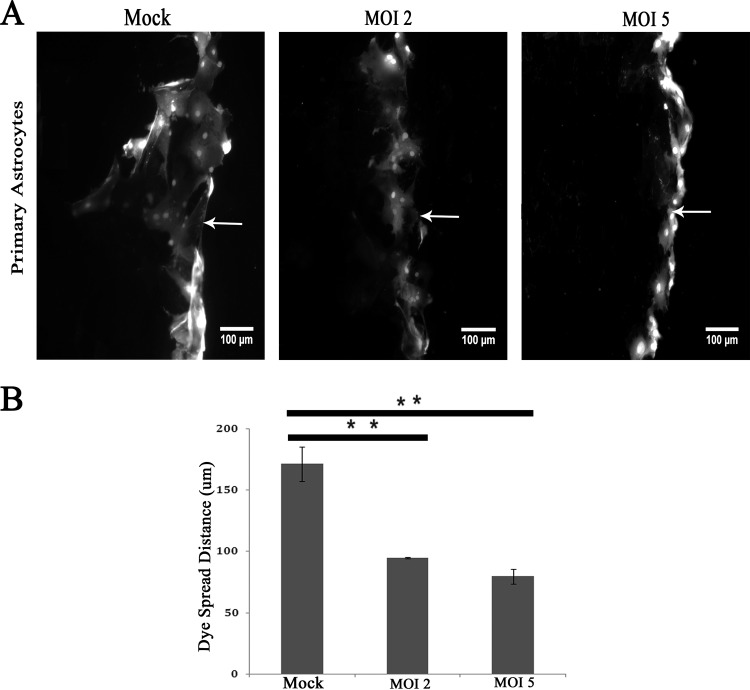
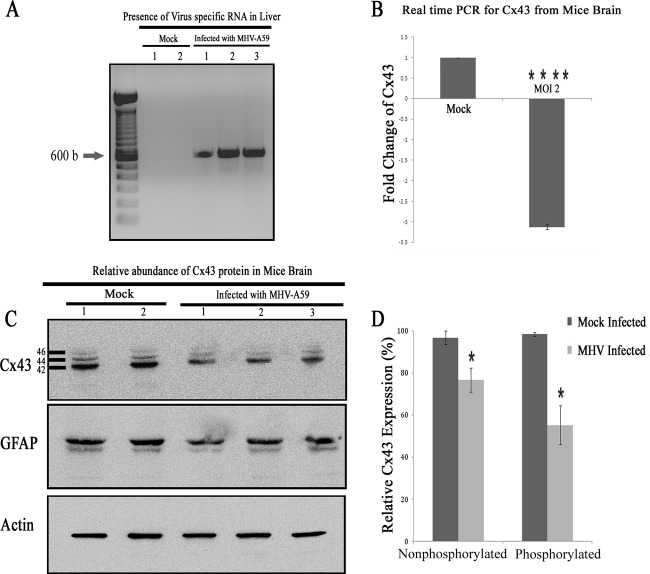
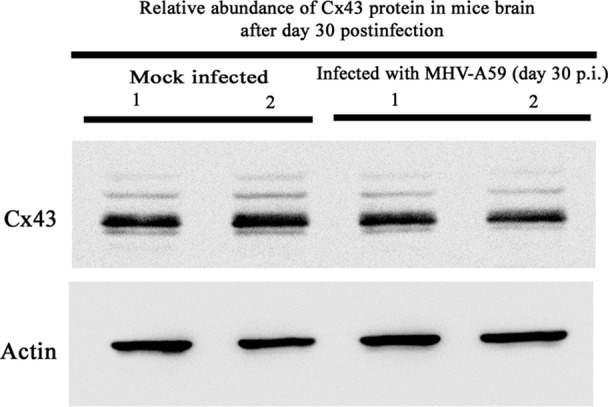
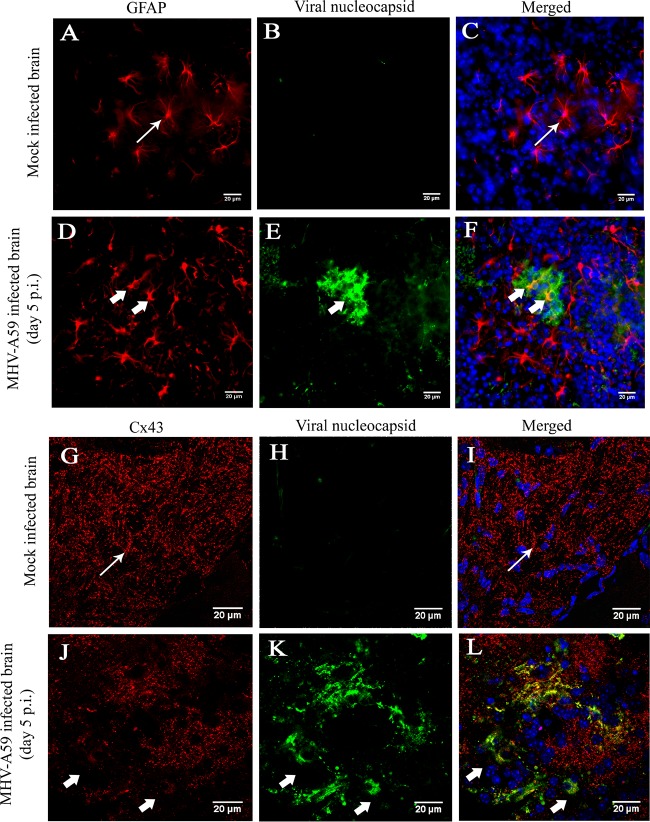
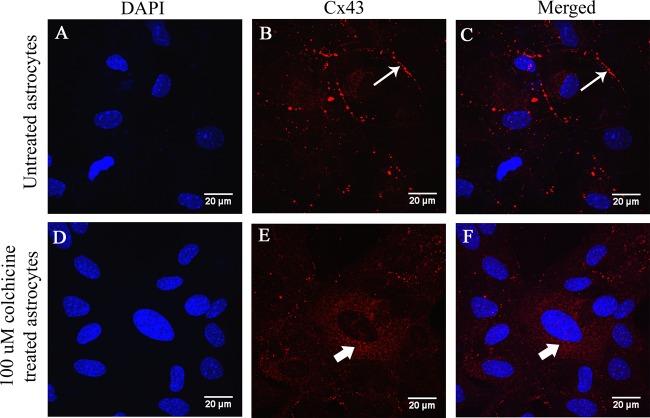
Gap junctions (GJs) form intercellular channels which directly connect the cytoplasm between neighboring cells to facilitate the transfer of ions and small molecules. GJs play a major role in the pathogenesis of infection-associated inflammation. Mutations of gap junction proteins, connexins (Cxs), cause dysmyelination and leukoencephalopathy. In multiple sclerosis (MS) patients and its animal model experimental autoimmune encephalitis (EAE), Cx43 was shown to be modulated in the central nervous system (CNS). The mechanism behind Cx43 alteration and its role in MS remains unexplored. Mouse hepatitis virus (MHV) infection-induced demyelination is one of the best-studied experimental animal models for MS. Our studies demonstrated that MHV infection downregulated Cx43 expression at protein and mRNA levels in vitro in primary astrocytes obtained from neonatal mouse brains. After infection, a significant amount of Cx43 was retained in endoplasmic reticulum/endoplasmic reticulum Golgi intermediate complex (ER/ERGIC) and GJ plaque formation was impaired at the cell surface, as evidenced by a reduction of the Triton X-100 insoluble fraction of Cx43. Altered trafficking and impairment of GJ plaque formation may cause the loss of functional channel formation in MHV-infected primary astrocytes, as demonstrated by a reduced number of dye-coupled cells after a scrape-loading Lucifer yellow dye transfer assay. Upon MHV infection, a significant downregulation of Cx43 was observed in the virus-infected mouse brain. This study demonstrates that astrocytic Cx43 expression and function can be modulated due to virus stress and can be an appropriate model to understand the basis of cellular mechanisms involved in the alteration of gap junction intercellular communication (GJIC) in CNS neuroinflammation.
Importance: We found that MHV infection leads to the downregulation of Cx43 in vivo in the CNS. In addition, results show that MHV infection impairs Cx43 expression in addition to gap junction communication in primary astrocytes. After infection, Cx43 did not traffic normally to the membrane to form gap junction plaques, and that could be the basis of reduced functional gap junction coupling between astrocytes. This is an important first step toward understanding how viruses affect Cx43 expression and trafficking at the cellular level. This may provide a basis for understanding how structural alterations of astrocytic gap junctions can disrupt gap junction communication between other CNS cells in altered CNS environments due to infection and inflammation. More specifically, alteration of Cx43 may be the basis of the destabilization of Cx47 in oligodendrocytes seen in and around inflammatory demyelinating plaques in MS patients.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures

Similar articles
-
Loss of Cx43-Mediated Functional Gap Junction Communication in Meningeal Fibroblasts Following Mouse Hepatitis Virus Infection.Mol Neurobiol. 2018 Aug;55(8):6558-6571. doi: 10.1007/s12035-017-0861-3. Epub 2018 Jan 11. Mol Neurobiol. 2018. PMID: 29327203 Free PMC article.
-
Microtubule-assisted altered trafficking of astrocytic gap junction protein connexin 43 is associated with depletion of connexin 47 during mouse hepatitis virus infection.J Biol Chem. 2017 Sep 8;292(36):14747-14763. doi: 10.1074/jbc.M117.786491. Epub 2017 May 31. J Biol Chem. 2017. PMID: 28566289 Free PMC article.
-
Human coronavirus OC43 infection remodels connexin 43-mediated gap junction intercellular communication in vitro.J Virol. 2024 Jul 23;98(7):e0047824. doi: 10.1128/jvi.00478-24. Epub 2024 May 31. J Virol. 2024. PMID: 38819132 Free PMC article.
-
Connexin 43/47 channels are important for astrocyte/ oligodendrocyte cross-talk in myelination and demyelination.J Biosci. 2018 Dec;43(5):1055-1068. doi: 10.1007/s12038-018-9811-0. J Biosci. 2018. PMID: 30541963 Free PMC article. Review.
-
Early disruption of glial communication via connexin gap junction in multiple sclerosis, Baló's disease and neuromyelitis optica.Neuropathology. 2015 Oct;35(5):469-80. doi: 10.1111/neup.12211. Epub 2015 May 28. Neuropathology. 2015. PMID: 26016402 Review.
Cited by
-
Amyloid-β regulates gap junction protein connexin 43 trafficking in cultured primary astrocytes.J Biol Chem. 2020 Oct 30;295(44):15097-15111. doi: 10.1074/jbc.RA120.013705. Epub 2020 Aug 31. J Biol Chem. 2020. PMID: 32868453 Free PMC article.
-
Loss of Cx43-Mediated Functional Gap Junction Communication in Meningeal Fibroblasts Following Mouse Hepatitis Virus Infection.Mol Neurobiol. 2018 Aug;55(8):6558-6571. doi: 10.1007/s12035-017-0861-3. Epub 2018 Jan 11. Mol Neurobiol. 2018. PMID: 29327203 Free PMC article.
-
Proline-Proline Dyad in the Fusion Peptide of the Murine β-Coronavirus Spike Protein's S2 Domain Modulates Its Neuroglial Tropism.Viruses. 2023 Jan 12;15(1):215. doi: 10.3390/v15010215. Viruses. 2023. PMID: 36680255 Free PMC article.
-
Identification of age-specific gene regulators of La Crosse virus neuroinvasion and pathogenesis.Nat Commun. 2023 May 18;14(1):2836. doi: 10.1038/s41467-023-37833-x. Nat Commun. 2023. PMID: 37202395 Free PMC article.
-
Connexin 43: An Interface Connecting Neuroinflammation to Depression.Molecules. 2023 Feb 15;28(4):1820. doi: 10.3390/molecules28041820. Molecules. 2023. PMID: 36838809 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
