

Evaluation of free modeling targets in CASP11 and ROLL
- PMID: 26677002
- PMCID: PMC5576440
- DOI: 10.1002/prot.24973
Evaluation of free modeling targets in CASP11 and ROLL
Abstract
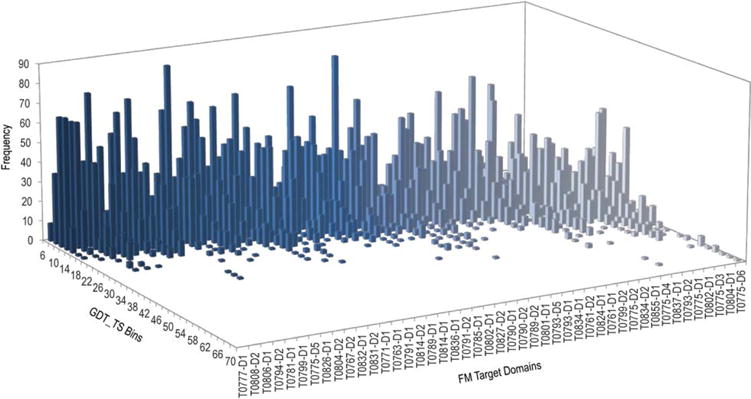
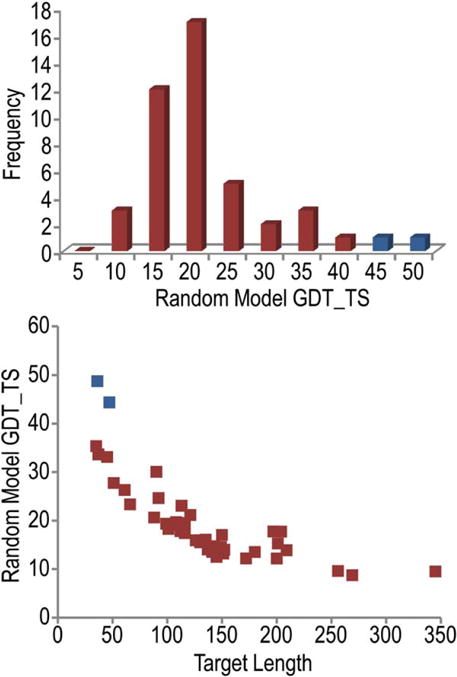
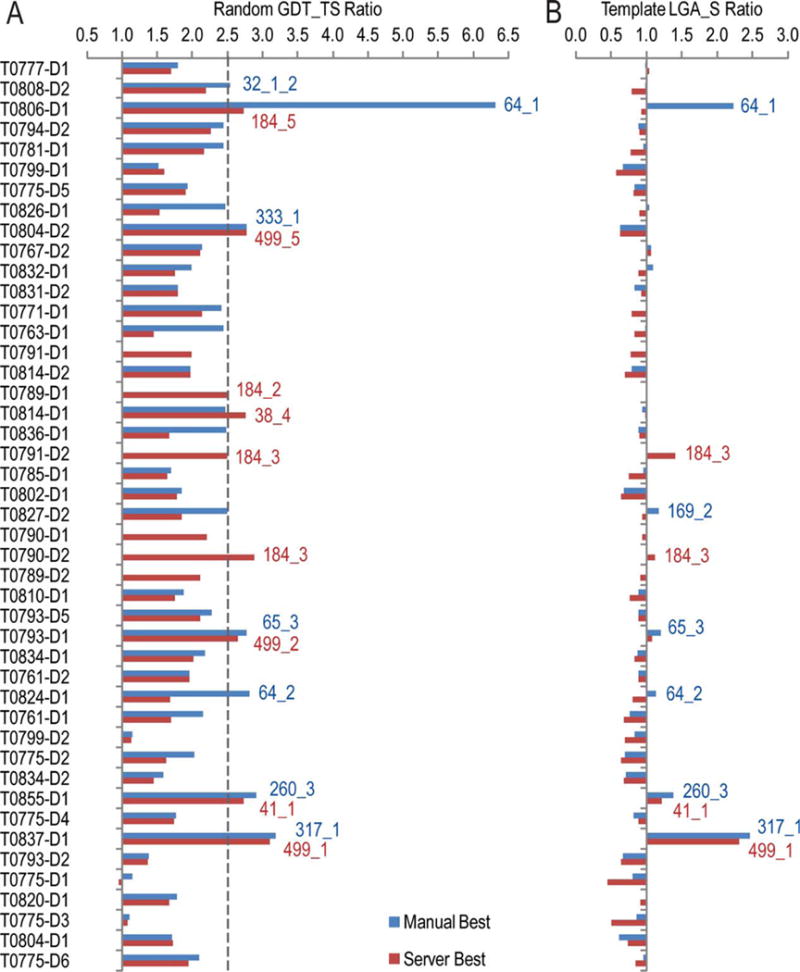
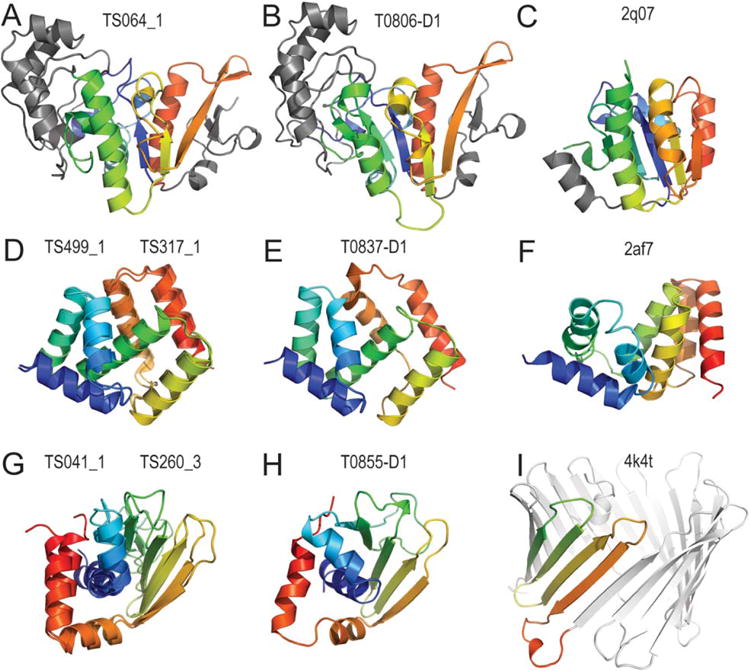
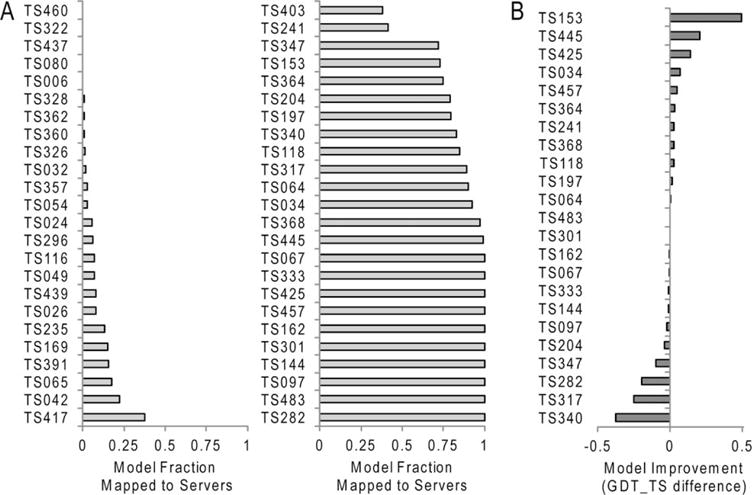
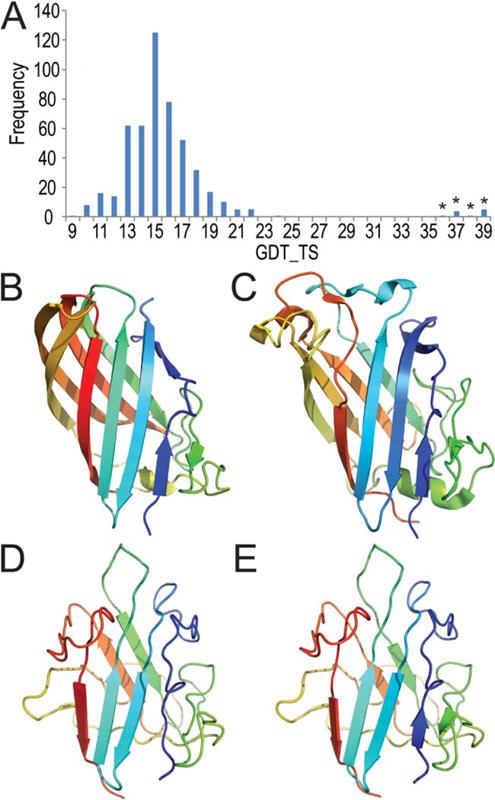
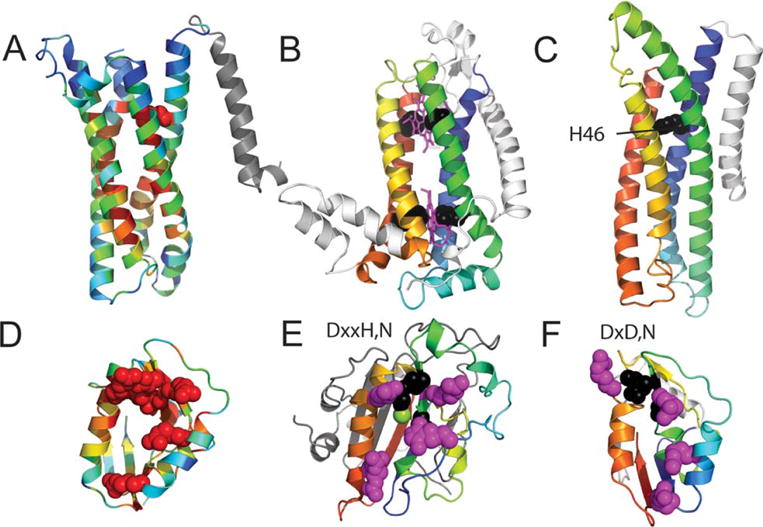
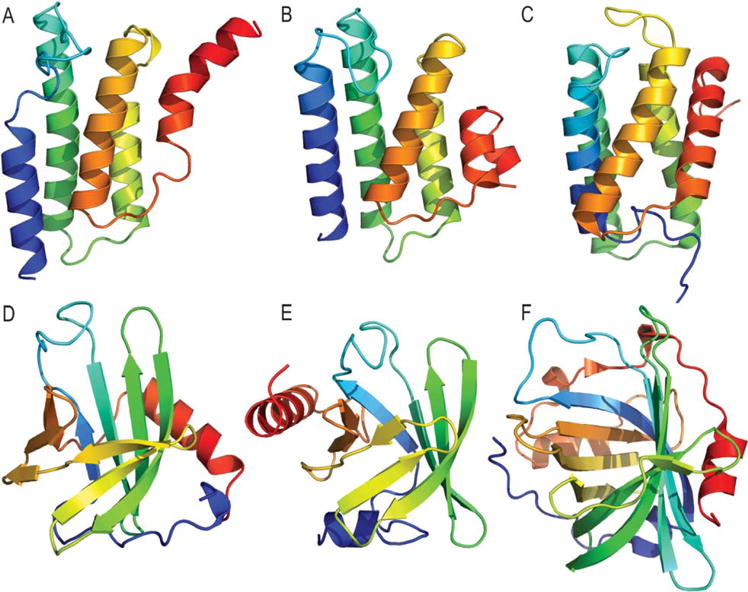
We present an assessment of 'template-free modeling' (FM) in CASP11and ROLL. Community-wide server performance suggested the use of automated scores similar to previous CASPs would provide a good system of evaluating performance, even in the absence of comprehensive manual assessment. The CASP11 FM category included several outstanding examples, including successful prediction by the Baker group of a 256-residue target (T0806-D1) that lacked sequence similarity to any existing template. The top server model prediction by Zhang's Quark, which was apparently selected and refined by several manual groups, encompassed the entire fold of target T0837-D1. Methods from the same two groups tended to dominate overall CASP11 FM and ROLL rankings. Comparison of top FM predictions with those from the previous CASP experiment revealed progress in the category, particularly reflected in high prediction accuracy for larger protein domains. FM prediction models for two cases were sufficient to provide functional insights that were otherwise not obtainable by traditional sequence analysis methods. Importantly, CASP11 abstracts revealed that alignment-based contact prediction methods brought about much of the CASP11 progress, producing both of the functionally relevant models as well as several of the other outstanding structure predictions. These methodological advances enabled de novo modeling of much larger domain structures than was previously possible and allowed prediction of functional sites. Proteins 2016; 84(Suppl 1):51-66. © 2015 Wiley Periodicals, Inc.
Keywords: CASP ROLL; CASP11; ab initio; alignment quality; domain structure; free modeling; protein fold prediction; protein structure; structure comparison.
© 2015 Wiley Periodicals, Inc.
Figures
References
-
- Lee J, Lee J, Sasaki TN, Sasai M, Seok C, Lee J. De novo protein structure prediction by dynamic fragment assembly and conformational space annealing. Proteins. 2011;79:2403–2417. - PubMed
-
- Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. - PubMed
-
- Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
