

Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling
- PMID: 26679539
- PMCID: PMC4699367
- DOI: 10.1186/s13059-015-0848-1
Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling
Abstract
Background: RNA levels detected at steady state are the consequence of multiple dynamic processes within the cell. In addition to synthesis and decay, transcripts undergo processing. Metabolic tagging with a nucleotide analog is one way of determining the relative contributions of synthesis, decay and conversion processes globally.
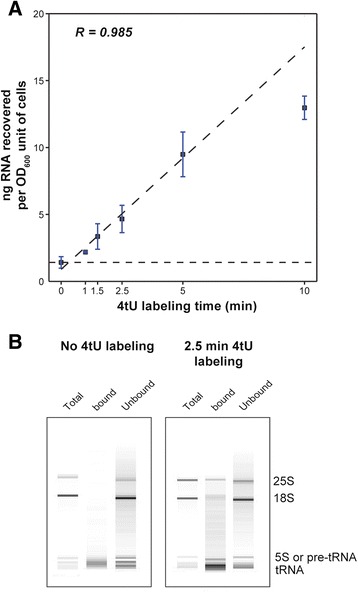
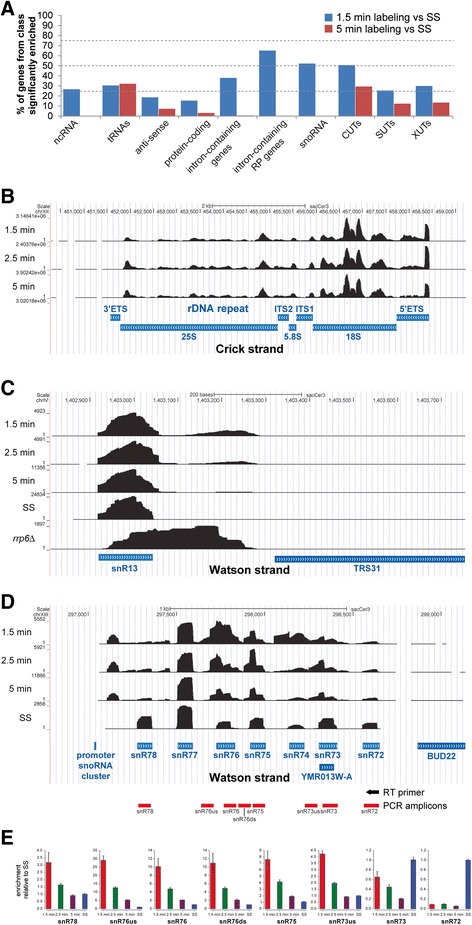
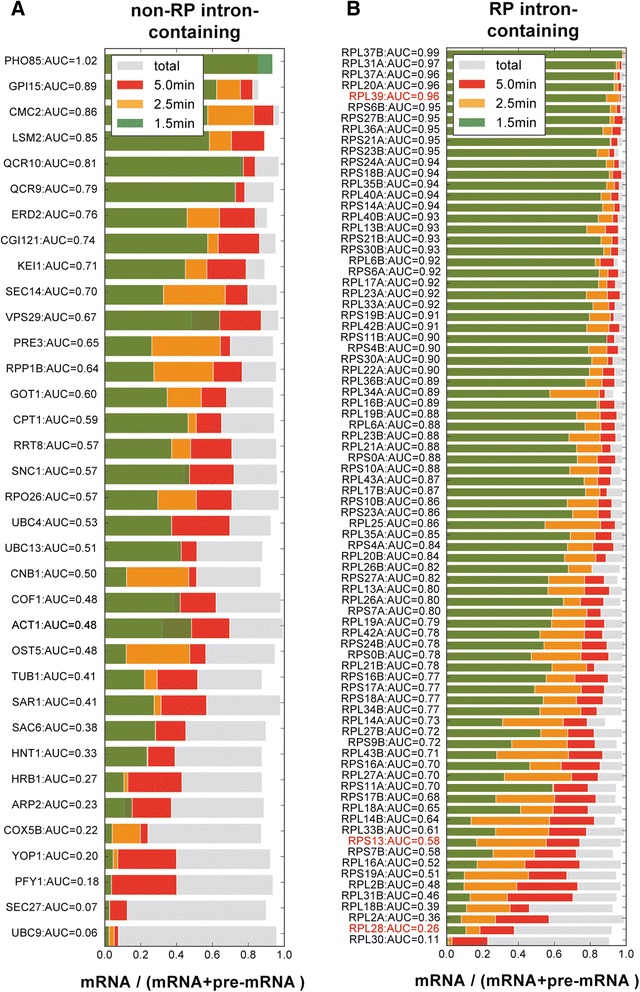
Results: By improving 4-thiouracil labeling of RNA in Saccharomyces cerevisiae we were able to isolate RNA produced during as little as 1 minute, allowing the detection of nascent pervasive transcription. Nascent RNA labeled for 1.5, 2.5 or 5 minutes was isolated and analyzed by reverse transcriptase-quantitative polymerase chain reaction and RNA sequencing. High kinetic resolution enabled detection and analysis of short-lived non-coding RNAs as well as intron-containing pre-mRNAs in wild-type yeast. From these data we measured the relative stability of pre-mRNA species with different high turnover rates and investigated potential correlations with sequence features.
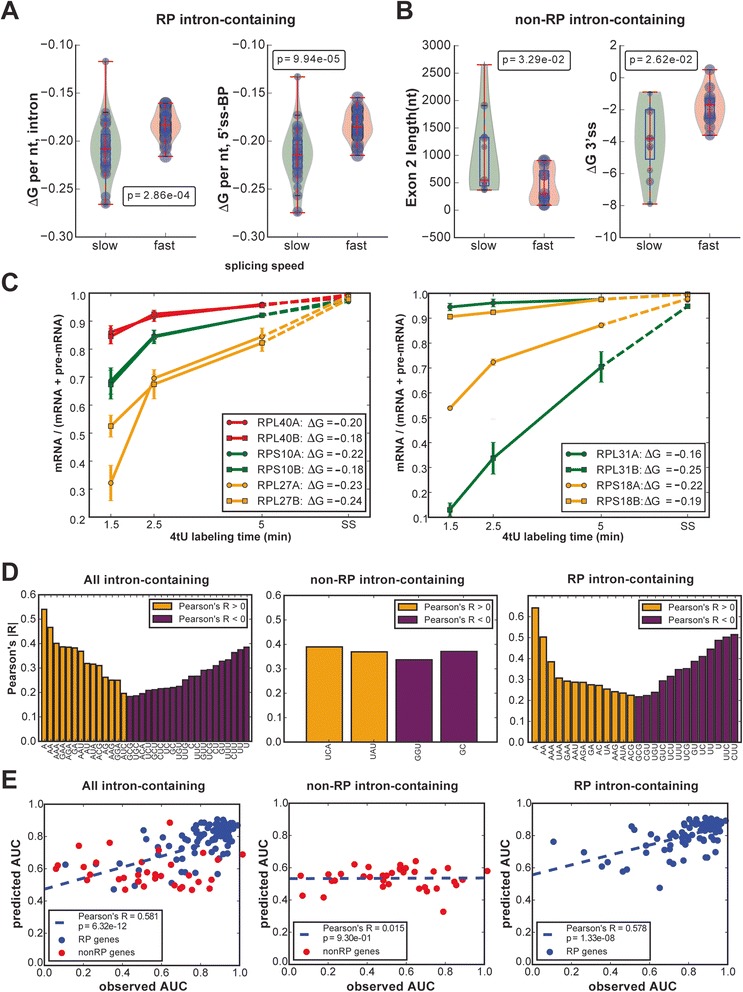
Conclusions: Our analysis of non-coding RNAs reveals a highly significant association between non-coding RNA stability, transcript length and predicted secondary structure. Our quantitative analysis of the kinetics of pre-mRNA splicing in yeast reveals that ribosomal protein transcripts are more efficiently spliced if they contain intron secondary structures that are predicted to be less stable. These data, in combination with previous results, indicate that there is an optimal range of stability of intron secondary structures that allows for rapid splicing.
Figures



References
-
- Yamashita A, Shichino Y, Yamamoto M. The long non-coding RNA world in yeasts. Biochim Biophys Acta. 2015. doi:10.1016/j.bbagrm.2015.08.003. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
