

Galiellalactone induces cell cycle arrest and apoptosis through the ATM/ATR pathway in prostate cancer cells
- PMID: 26683224
- PMCID: PMC4826221
- DOI: 10.18632/oncotarget.6606
Galiellalactone induces cell cycle arrest and apoptosis through the ATM/ATR pathway in prostate cancer cells
Abstract
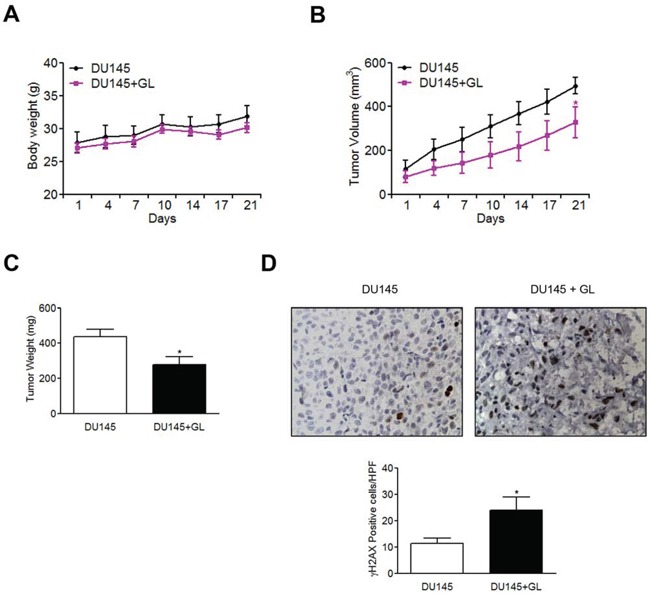
Galiellalactone (GL) is a fungal metabolite that presents antitumor activities on prostate cancer in vitro and in vivo. In this study we show that GL induced cell cycle arrest in G2/M phase, caspase-dependent apoptosis and also affected the microtubule organization and migration ability in DU145 cells. GL did not induce double strand DNA break but activated the ATR and ATM-mediated DNA damage response (DDR) inducing CHK1, H2AX phosphorylation (fH2AX) and CDC25C downregulation. Inhibition of the ATM/ATR activation with caffeine reverted GL-induced G2/M cell cycle arrest, apoptosis and DNA damage measured by fH2AX. In contrast, UCN-01, a CHK1 inhibitor, prevented GL-induced cell cycle arrest but enhanced apoptosis in DU145 cells. Furthermore, we found that GL did not increase the levels of intracellular ROS, but the antioxidant N-acetylcysteine (NAC) completely prevented the effects of GL on fH2AX, G2/M cell cycle arrest and apoptosis. In contrast to NAC, other antioxidants such as ambroxol and EGCG did not interfere with the activity of GL on cell cycle. GL significantly suppressed DU145 xenograft growth in vivo and induced the expression of fH2AX in the tumors. These findings identify for the first time that GL activates DDR in prostate cancer.
Keywords: ATM/ATR; CHK1; cancer; cell cycle; galiellalactone.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








References
-
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA. 2015;65:87–108. - PubMed
-
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nature reviews Cancer. 2001;1:34–45. - PubMed
-
- Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clinical cancer research. 2006;12:1665–1671. - PubMed
-
- Katzenwadel A, Wolf P. Androgen deprivation of prostate cancer: Leading to a therapeutic dead end. Cancer letters. 2015 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
