

Dimethyl Fumarate Inhibits the Nuclear Factor κB Pathway in Breast Cancer Cells by Covalent Modification of p65 Protein
- PMID: 26683377
- PMCID: PMC4751401
- DOI: 10.1074/jbc.M115.679704
Dimethyl Fumarate Inhibits the Nuclear Factor κB Pathway in Breast Cancer Cells by Covalent Modification of p65 Protein
Abstract
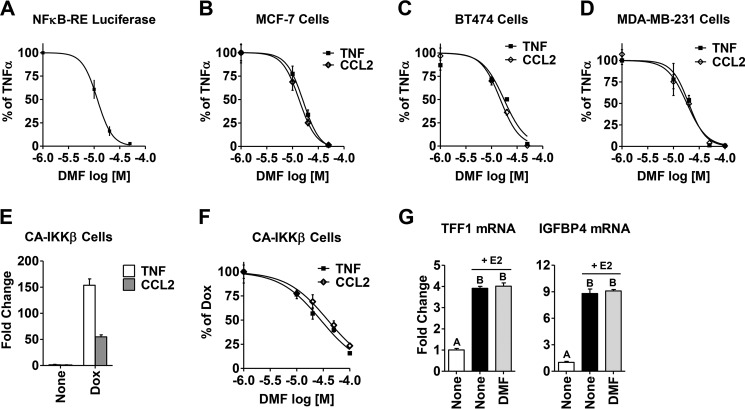
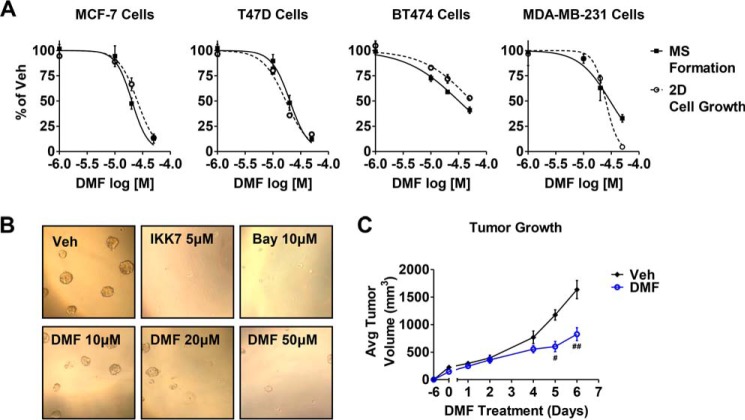
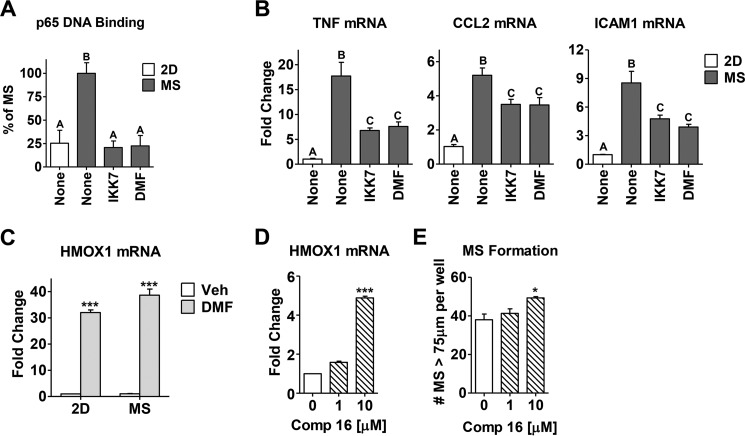
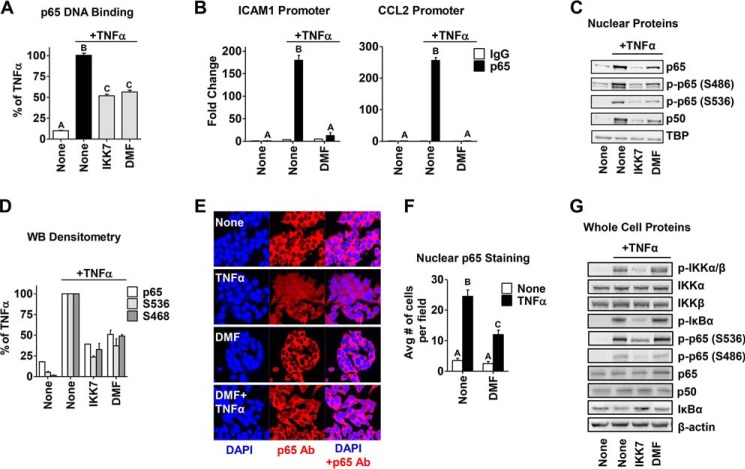
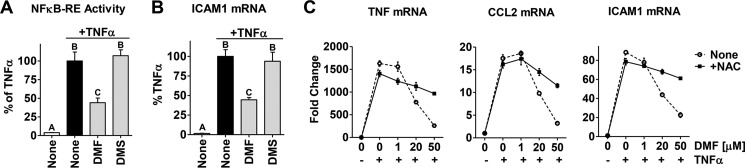
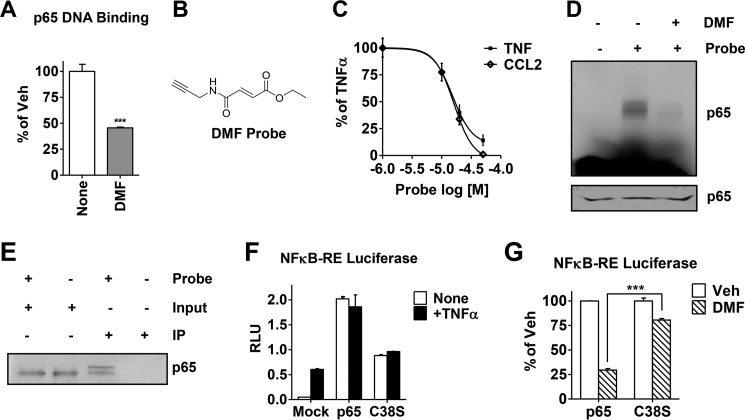
In breast tumors, activation of the nuclear factor κB (NFκB) pathway promotes survival, migration, invasion, angiogenesis, stem cell-like properties, and resistance to therapy--all phenotypes of aggressive disease where therapy options remain limited. Adding an anti-inflammatory/anti-NFκB agent to breast cancer treatment would be beneficial, but no such drug is approved as either a monotherapy or adjuvant therapy. To address this need, we examined whether dimethyl fumarate (DMF), an anti-inflammatory drug already in clinical use for multiple sclerosis, can inhibit the NFκB pathway. We found that DMF effectively blocks NFκB activity in multiple breast cancer cell lines and abrogates NFκB-dependent mammosphere formation, indicating that DMF has anti-cancer stem cell properties. In addition, DMF inhibits cell proliferation and significantly impairs xenograft tumor growth. Mechanistically, DMF prevents p65 nuclear translocation and attenuates its DNA binding activity but has no effect on upstream proteins in the NFκB pathway. Dimethyl succinate, the inactive analog of DMF that lacks the electrophilic double bond of fumarate, is unable to inhibit NFκB activity. Also, the cell-permeable thiol N-acetyl l-cysteine, reverses DMF inhibition of the NFκB pathway, supporting the notion that the electrophile, DMF, acts via covalent modification. To determine whether DMF interacts directly with p65, we synthesized and used a novel chemical probe of DMF by incorporating an alkyne functionality and found that DMF covalently modifies p65, with cysteine 38 being essential for the activity of DMF. These results establish DMF as an NFκB inhibitor with anti-tumor activity that may add therapeutic value in the treatment of aggressive breast cancers.
Keywords: NFκB; breast cancer; chemical modification; covalent modification; cysteine; dimethyl fumarate; drug action; inflammation; inhibitor; mammosphere.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 - PubMed
-
- Karin M., Cao Y., Greten F. R., and Li Z. W. (2002) NF-κB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2, 301–310 - PubMed
-
- Kim H. J., Hawke N., and Baldwin A. S. (2006) NF-κB and IKK as therapeutic targets in cancer. Cell Death Differ. 13, 738–747 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
