

Aspartate β-hydroxylase modulates cellular senescence through glycogen synthase kinase 3β in hepatocellular carcinoma
- PMID: 26683595
- PMCID: PMC4805474
- DOI: 10.1002/hep.28411
Aspartate β-hydroxylase modulates cellular senescence through glycogen synthase kinase 3β in hepatocellular carcinoma
Abstract
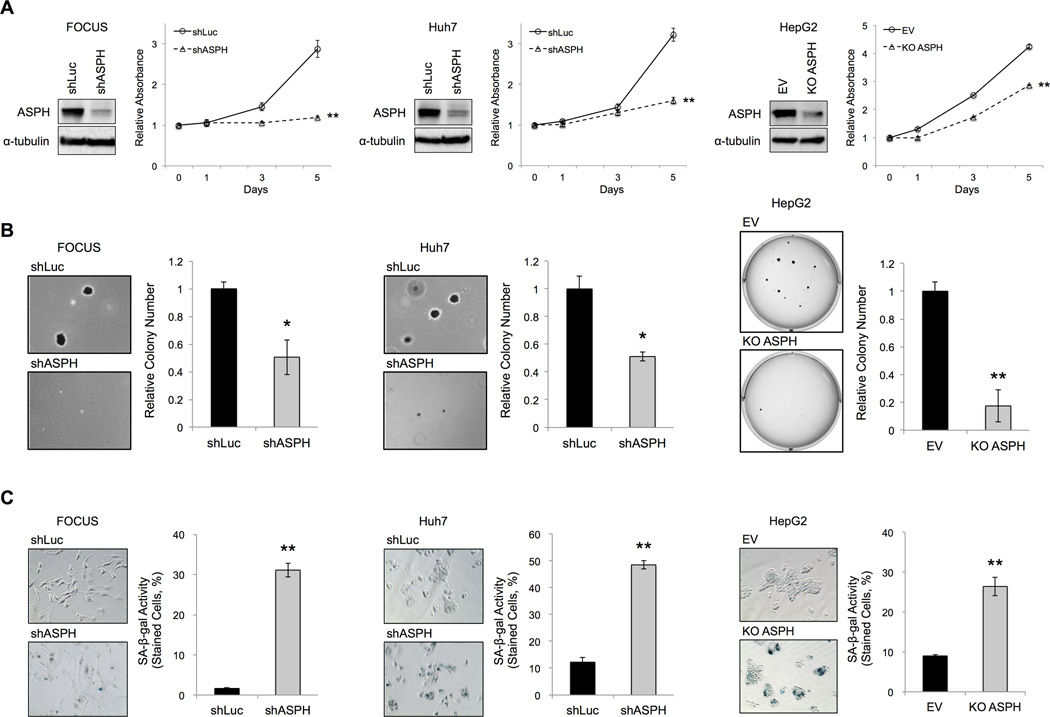
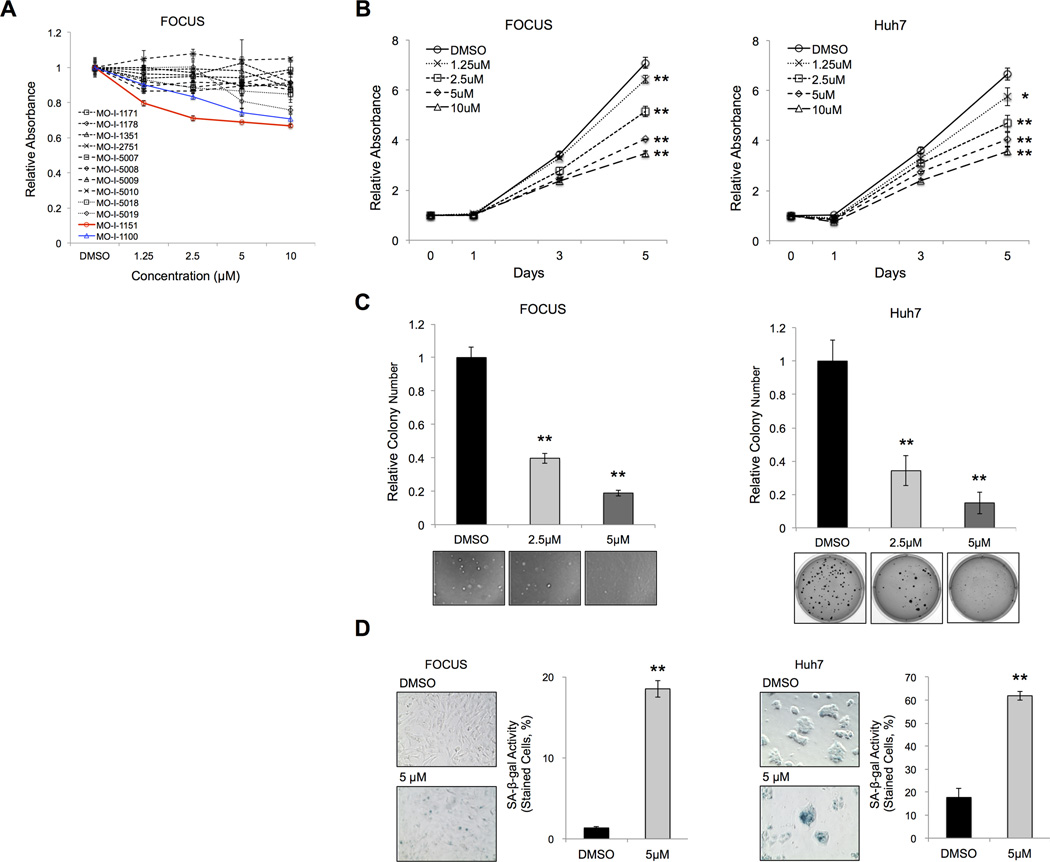
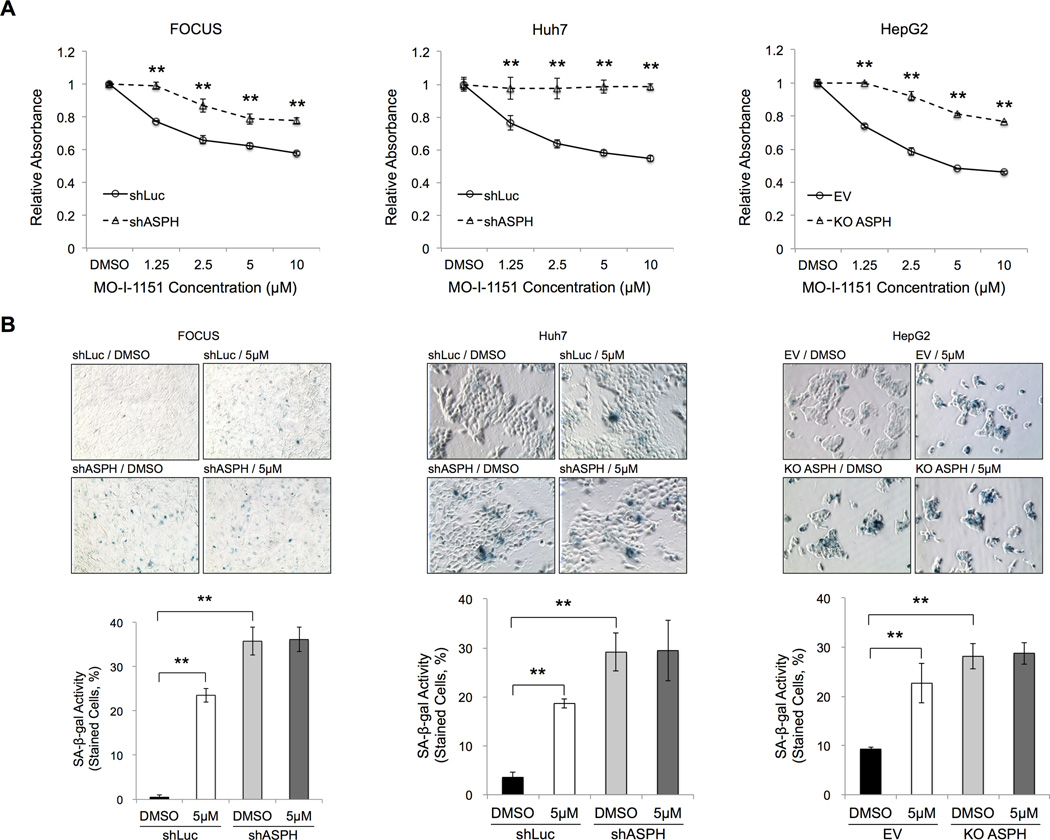
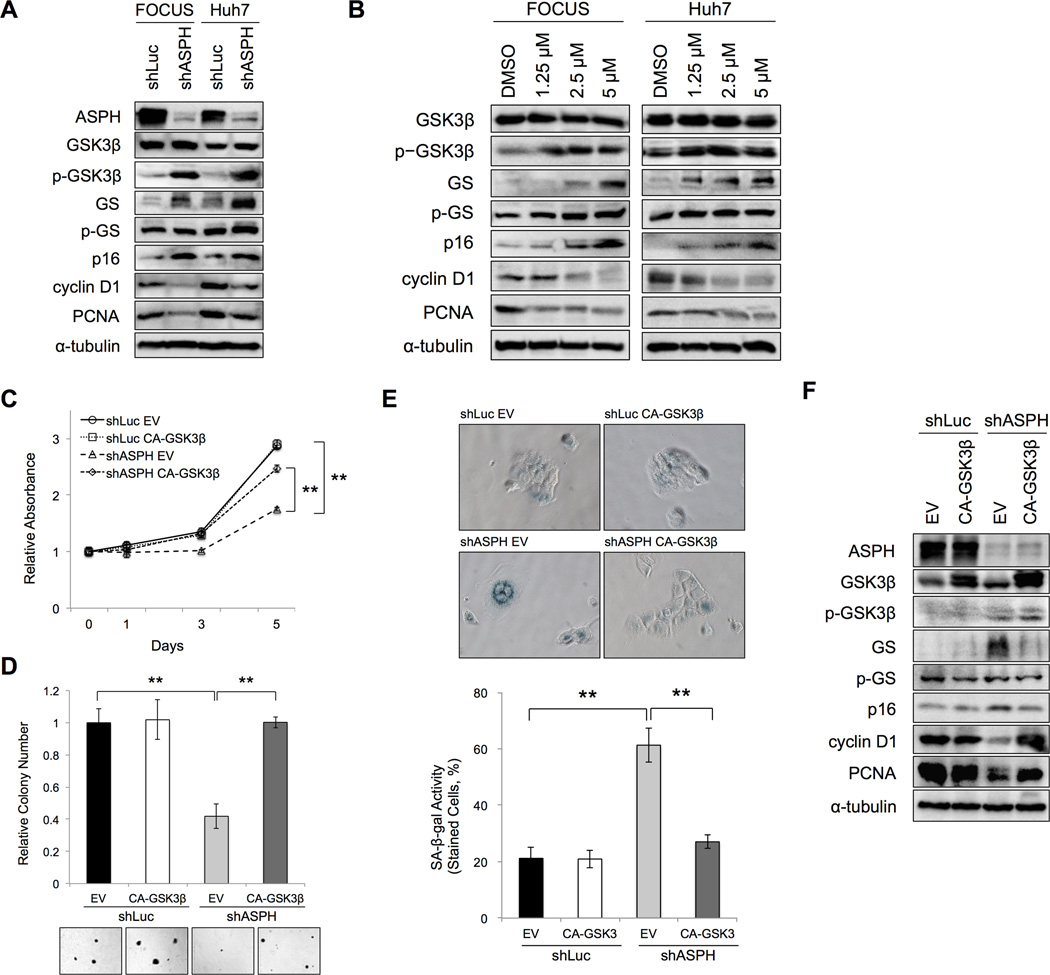
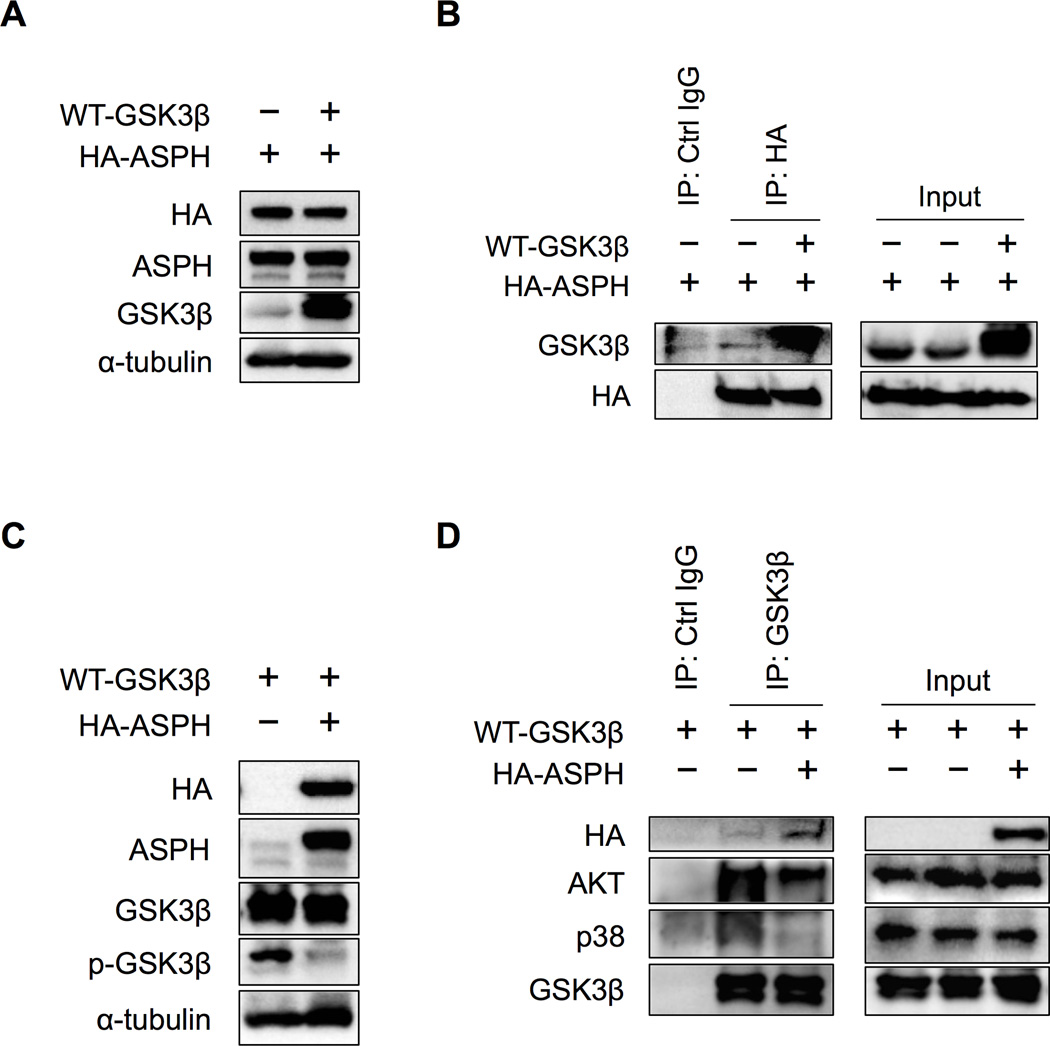
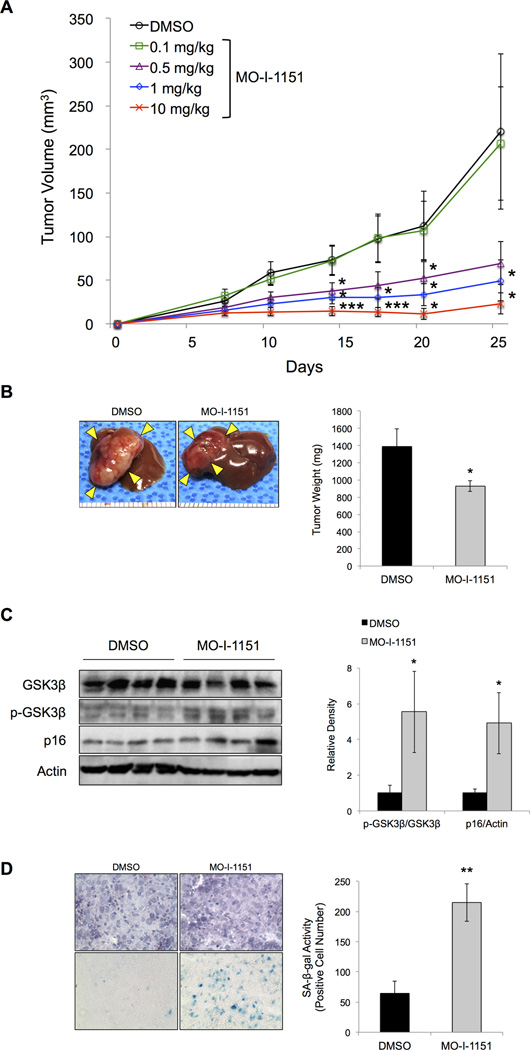
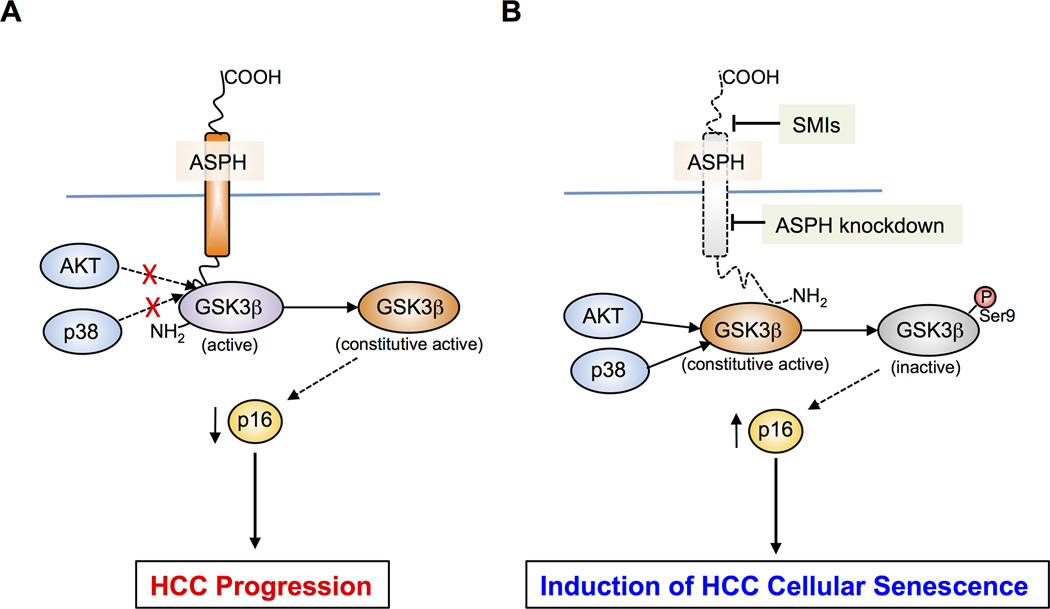
Aspartate β-hydroxylase (ASPH) is an enzyme overexpressed in human hepatocellular carcinoma (HCC) tumors that participates in the malignant transformation process. We determined if ASPH was a therapeutic target by exerting effects on cellular senescence to retard HCC progression. ASPH knockdown or knockout was achieved by short hairpin RNAs or the CRISPR/Cas9 system, respectively, whereas enzymatic inhibition was rendered by a potent second-generation small molecule inhibitor of ASPH. Alterations of cell proliferation, colony formation, and cellular senescence were evaluated in human HCC cell lines. The potential mechanisms for activating cellular senescence were explored using murine subcutaneous and orthotopic xenograft models. Inhibition of ASPH expression and enzymatic activity significantly reduced cell proliferation and colony formation but induced tumor cell senescence. Following inhibition of ASPH activity, phosphorylation of glycogen synthase kinase 3β and p16 expression were increased to promote senescence, whereas cyclin D1 and proliferating cell nuclear antigen were decreased to reduce cell proliferation. The mechanisms involved demonstrate that ASPH binds to glycogen synthase kinase 3β and inhibits its subsequent interactions with protein kinase B and p38 upstream kinases as shown by coimmunoprecipitation. In vivo experiments demonstrated that small molecule inhibitor treatment of HCC bearing mice resulted in significant dose-dependent reduced tumor growth, induced phosphorylation of glycogen synthase kinase 3β, enhanced p16 expression in tumor cells, and promoted cellular senescence.
Conclusions: We have identified a new mechanism that promotes HCC growth and progression by modulating senescence of tumor cells; these findings suggest that ASPH enzymatic activity is a novel therapeutic target for HCC.
© 2015 by the American Association for the Study of Liver Diseases.
Conflict of interest statement
Figures
References
-
- Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001;2:533–543. - PubMed
-
- El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. - PubMed
-
- Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol. 2013;10:34–42. - PubMed
-
- Jia S, VanDusen WJ, Diehl RE, Kohl NE, Dixon RA, Elliston KO, Stern AM, et al. cDNA cloning and expression of bovine aspartyl (asparaginyl) beta-hydroxylase. J Biol Chem. 1992;267:14322–14327. - PubMed
-
- Engel J. EGF-like domains in extracellular matrix proteins: localized signals for growth and differentiation? FEBS Lett. 1989;251:1–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
