

The Constrained Maximal Expression Level Owing to Haploidy Shapes Gene Content on the Mammalian X Chromosome
- PMID: 26685068
- PMCID: PMC4686125
- DOI: 10.1371/journal.pbio.1002315
The Constrained Maximal Expression Level Owing to Haploidy Shapes Gene Content on the Mammalian X Chromosome
Abstract
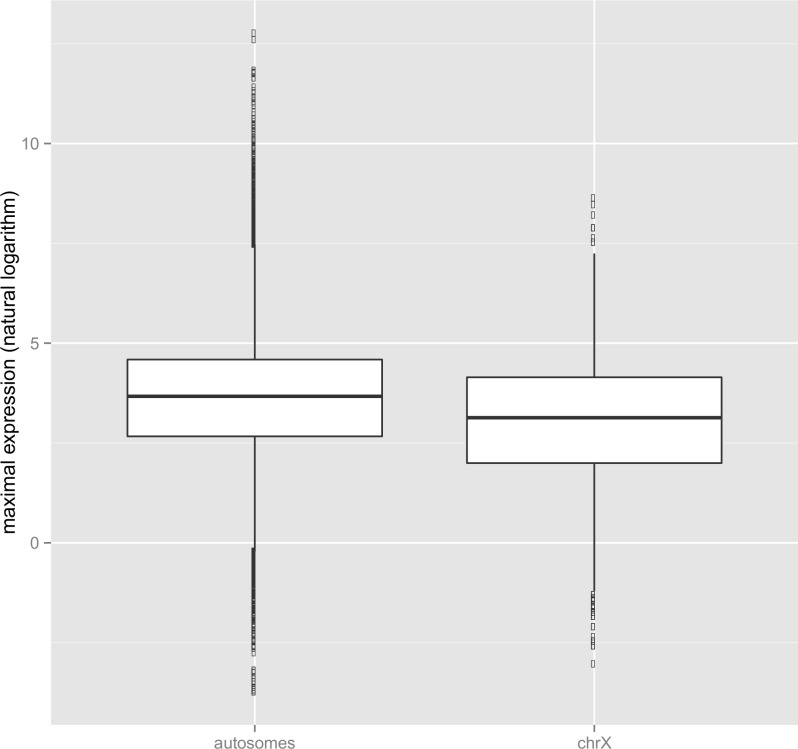
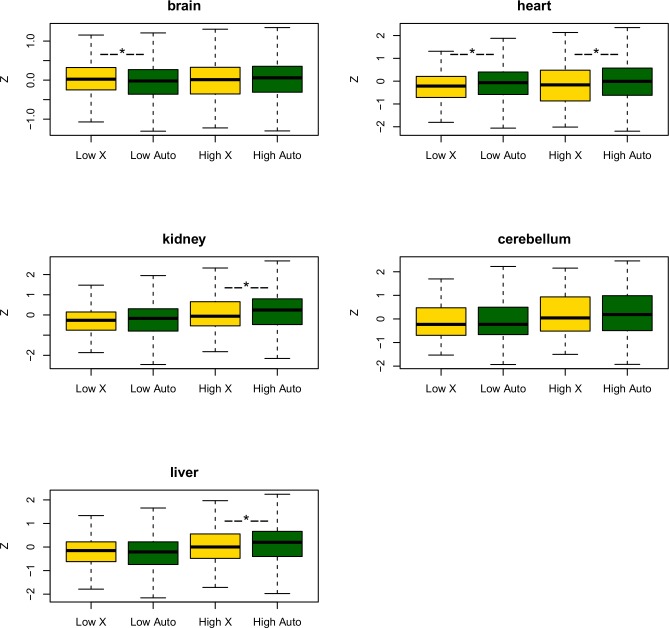
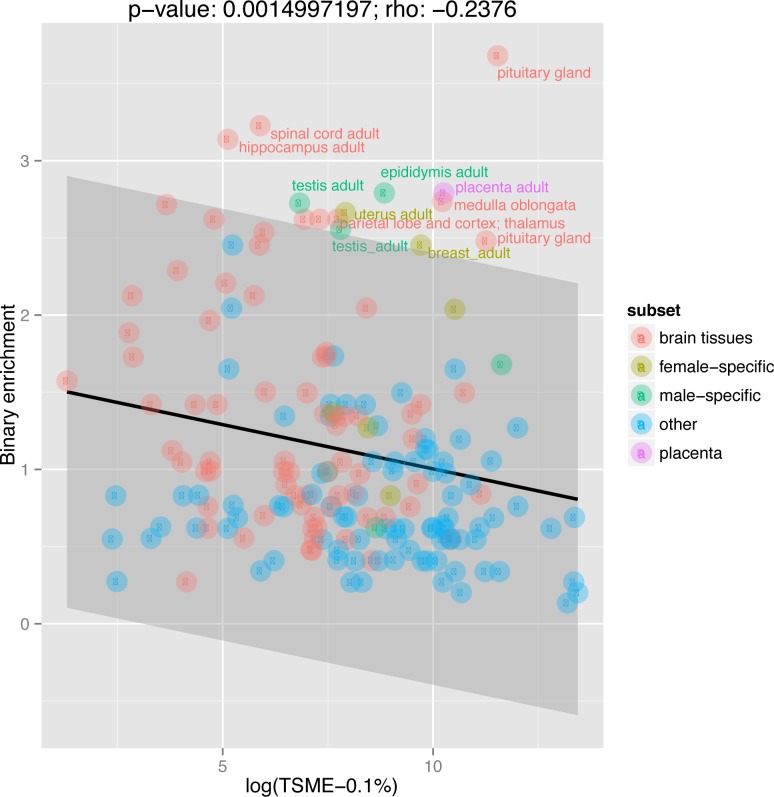
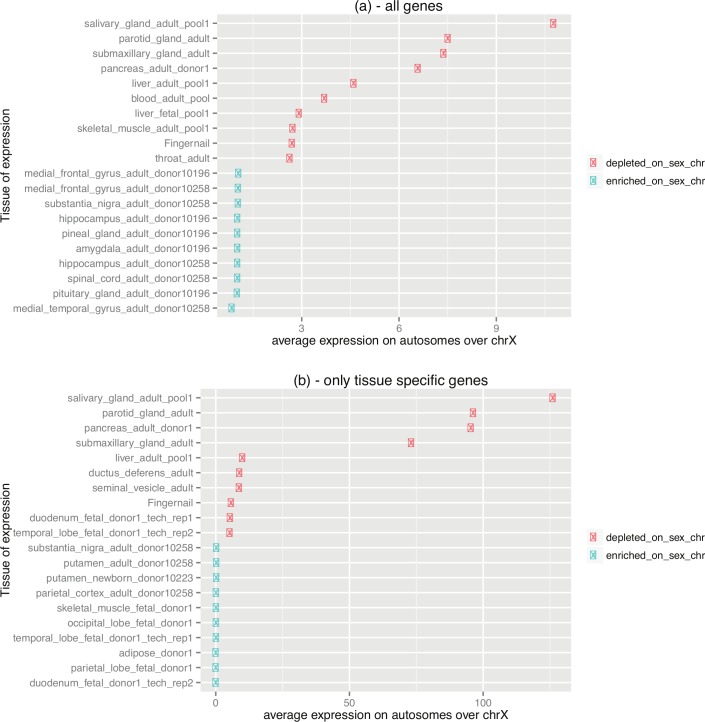
X chromosomes are unusual in many regards, not least of which is their nonrandom gene content. The causes of this bias are commonly discussed in the context of sexual antagonism and the avoidance of activity in the male germline. Here, we examine the notion that, at least in some taxa, functionally biased gene content may more profoundly be shaped by limits imposed on gene expression owing to haploid expression of the X chromosome. Notably, if the X, as in primates, is transcribed at rates comparable to the ancestral rate (per promoter) prior to the X chromosome formation, then the X is not a tolerable environment for genes with very high maximal net levels of expression, owing to transcriptional traffic jams. We test this hypothesis using The Encyclopedia of DNA Elements (ENCODE) and data from the Functional Annotation of the Mammalian Genome (FANTOM5) project. As predicted, the maximal expression of human X-linked genes is much lower than that of genes on autosomes: on average, maximal expression is three times lower on the X chromosome than on autosomes. Similarly, autosome-to-X retroposition events are associated with lower maximal expression of retrogenes on the X than seen for X-to-autosome retrogenes on autosomes. Also as expected, X-linked genes have a lesser degree of increase in gene expression than autosomal ones (compared to the human/Chimpanzee common ancestor) if highly expressed, but not if lowly expressed. The traffic jam model also explains the known lower breadth of expression for genes on the X (and the Z of birds), as genes with broad expression are, on average, those with high maximal expression. As then further predicted, highly expressed tissue-specific genes are also rare on the X and broadly expressed genes on the X tend to be lowly expressed, both indicating that the trend is shaped by the maximal expression level not the breadth of expression per se. Importantly, a limit to the maximal expression level explains biased tissue of expression profiles of X-linked genes. Tissues whose tissue-specific genes are very highly expressed (e.g., secretory tissues, tissues abundant in structural proteins) are also tissues in which gene expression is relatively rare on the X chromosome. These trends cannot be fully accounted for in terms of alternative models of biased expression. In conclusion, the notion that it is hard for genes on the Therian X to be highly expressed, owing to transcriptional traffic jams, provides a simple yet robustly supported rationale of many peculiar features of X's gene content, gene expression, and evolution.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
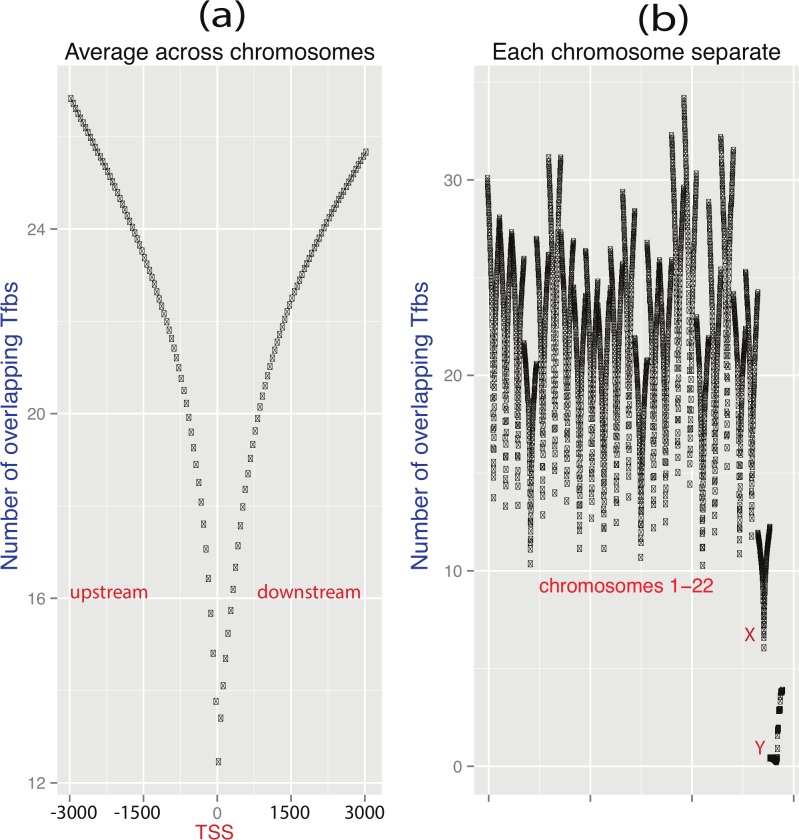
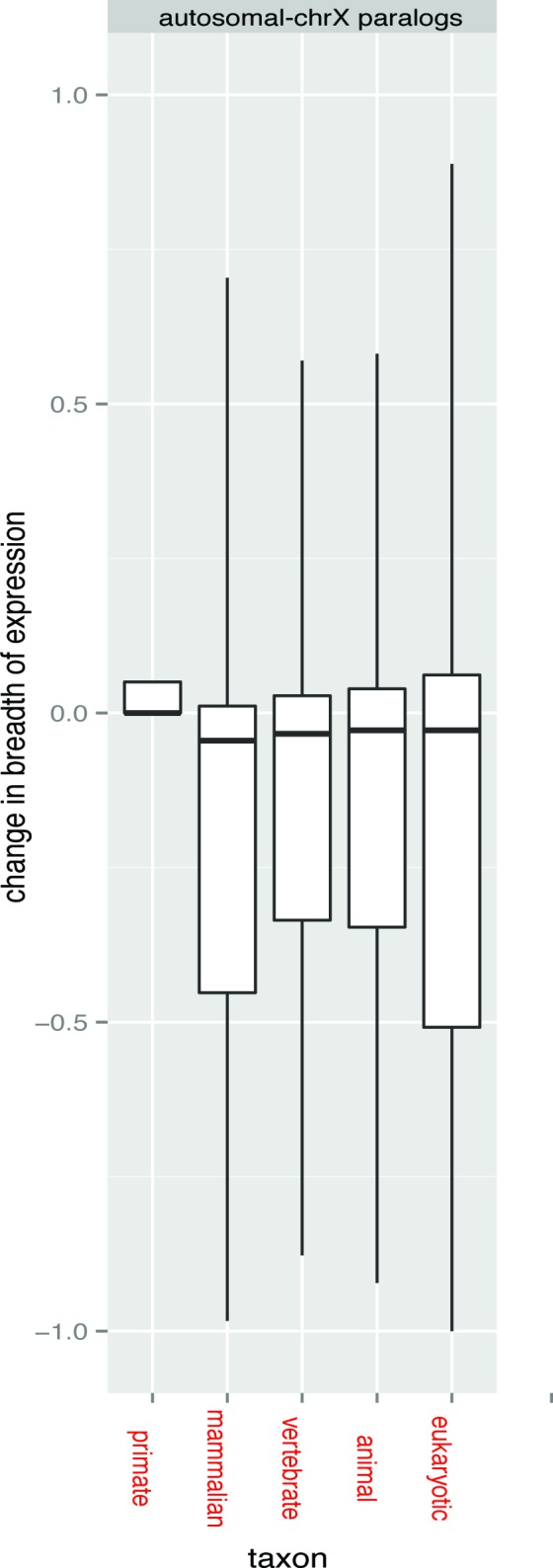
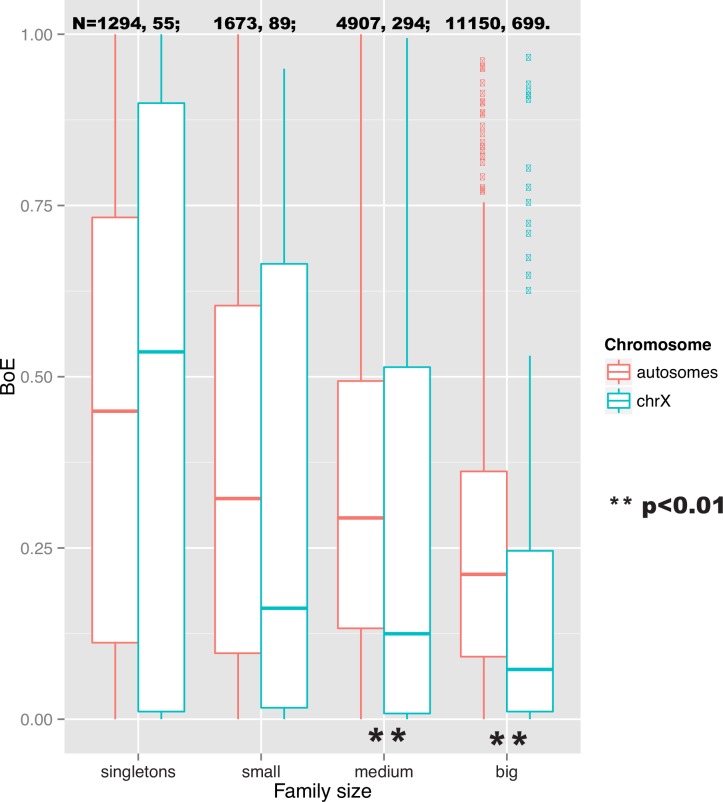
References
-
- Ohno DS. Sex Chromosomes and Sex-Linked Genes. Berlin, Heidelberg, New York: Springer-Verlag; 1967.
-
- Charlesworth B, Coyne JA, Barton NH. The Relative Rates of Evolution of Sex Chromosomes and Autosomes. Am Nat. 1987;130(1):113–46.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
