

Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome
- PMID: 26685159
- PMCID: PMC4743690
- DOI: 10.1093/hmg/ddv507
Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome
Abstract
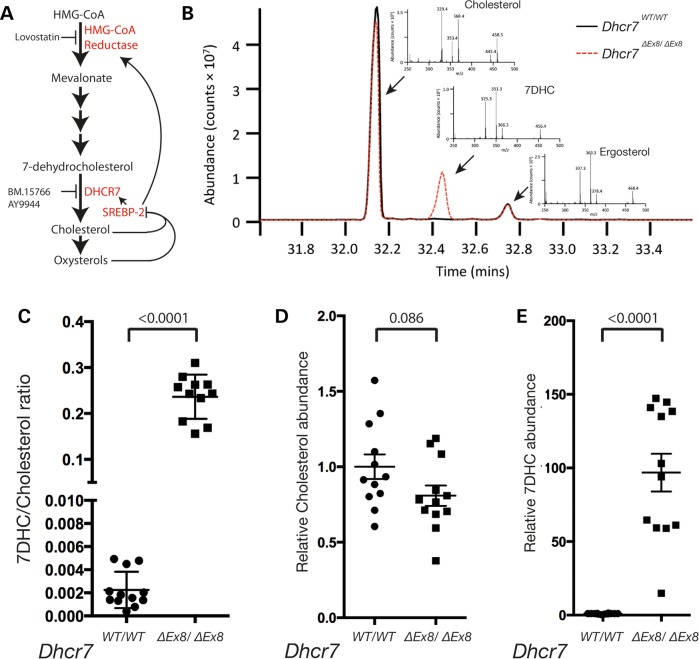
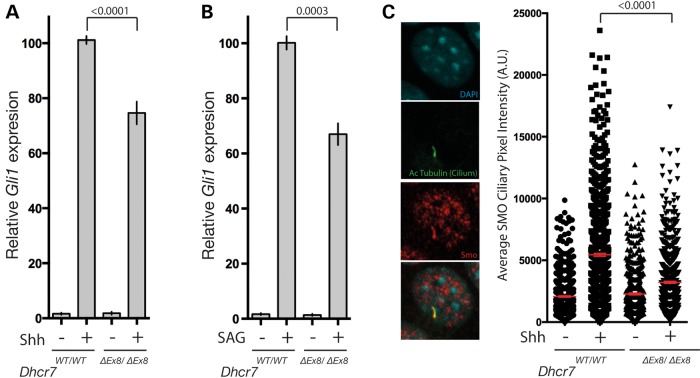
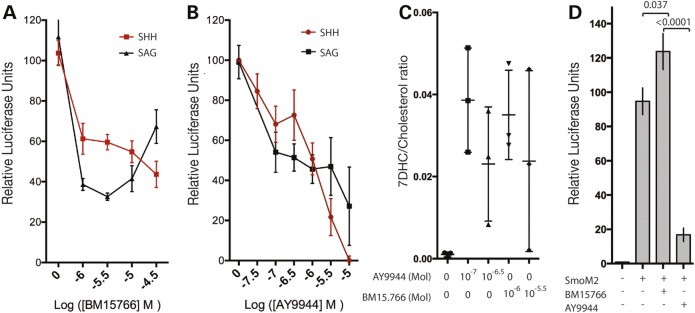
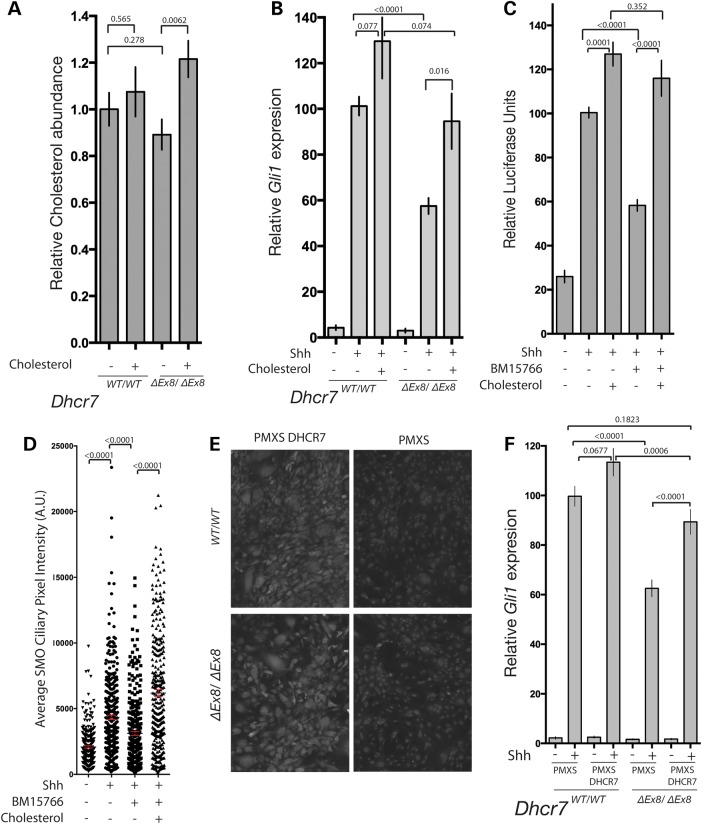
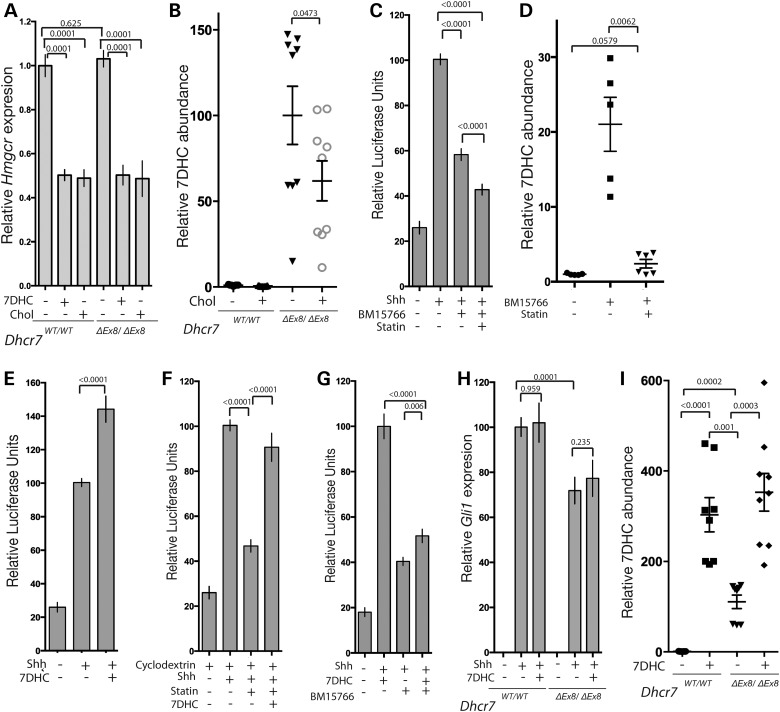
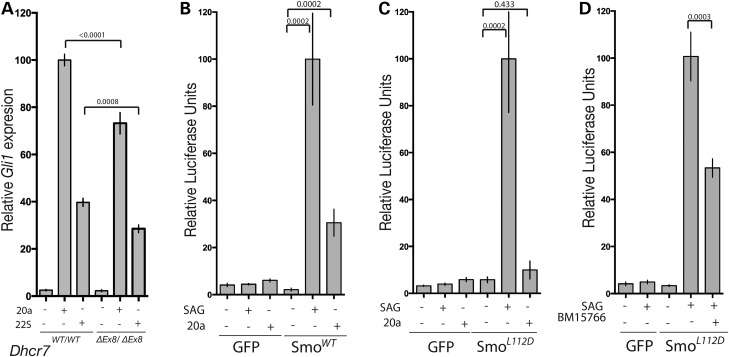
Smith-Lemli-Opitz syndrome (SLOS) is a common autosomal-recessive disorder that results from mutations in the gene encoding the cholesterol biosynthetic enzyme 7-dehydrocholesterol reductase (DHCR7). Impaired DHCR7 function is associated with a spectrum of congenital malformations, intellectual impairment, epileptiform activity and autism spectrum disorder. Biochemically, there is a deficit in cholesterol and an accumulation of its metabolic precursor 7-dehydrocholesterol (7DHC) in developing tissues. Morphological abnormalities in SLOS resemble those seen in congenital Sonic Hedgehog (SHH)-deficient conditions, leading to the proposal that the pathogenesis of SLOS is mediated by aberrant SHH signalling. SHH signalling is transduced through the transmembrane protein Smoothened (SMO), which localizes to the primary cilium of a cell on activation and is both positively and negatively regulated by sterol molecules derived from cholesterol biosynthesis. One proposed mechanism of SLOS involves SMO dysregulation by altered sterol levels, but the salient sterol species has not been identified. Here, we clarify the relationship between disrupted cholesterol metabolism and reduced SHH signalling in SLOS by modelling the disorder in vitro. Our results indicate that a deficit in cholesterol, as opposed to an accumulation of 7DHC, impairs SMO activation and its localization to the primary cilium.
© The Author 2015. Published by Oxford University Press.
Figures
References
-
- Nowaczyk M.J., Zeesman S., Waye J.S., Douketis J.D. (2004) Incidence of Smith-Lemli-Opitz syndrome in Canada: results of three-year population surveillance. J. Pediatr., 145, 530–535. - PubMed
-
- Tint G.S., Irons M., Elias E.R., Batta A.K., Frieden R., Chen T.S., Salen G. (1994) Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med., 330, 107–113. - PubMed
-
- Nowaczyk M.J., McCaughey D., Whelan D.T., Porter F.D. (2001) Incidence of Smith-Lemli-Opitz syndrome in Ontario, Canada. Am. J. Med. Genet., 102, 18–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
