

Peroxisomes in brain development and function
- PMID: 26686055
- PMCID: PMC4880039
- DOI: 10.1016/j.bbamcr.2015.12.005
Peroxisomes in brain development and function
Abstract
Peroxisomes contain numerous enzymatic activities that are important for mammalian physiology. Patients lacking either all peroxisomal functions or a single enzyme or transporter function typically develop severe neurological deficits, which originate from aberrant development of the brain, demyelination and loss of axonal integrity, neuroinflammation or other neurodegenerative processes. Whilst correlating peroxisomal properties with a compilation of pathologies observed in human patients and mouse models lacking all or individual peroxisomal functions, we discuss the importance of peroxisomal metabolites and tissue- and cell type-specific contributions to the observed brain pathologies. This enables us to deconstruct the local and systemic contribution of individual metabolic pathways to specific brain functions. We also review the recently discovered variability of pathological symptoms in cases with unexpectedly mild presentation of peroxisome biogenesis disorders. Finally, we explore the emerging evidence linking peroxisomes to more common neurological disorders such as Alzheimer's disease, autism and amyotrophic lateral sclerosis.
Keywords: D-bifunctional protein deficiency; Lipid metabolism; Plasmalogen; Rhizomelic chondrodysplasia punctata; X-linked adrenoleukodystrophy; Zellweger spectrum disorder.
Copyright © 2015 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
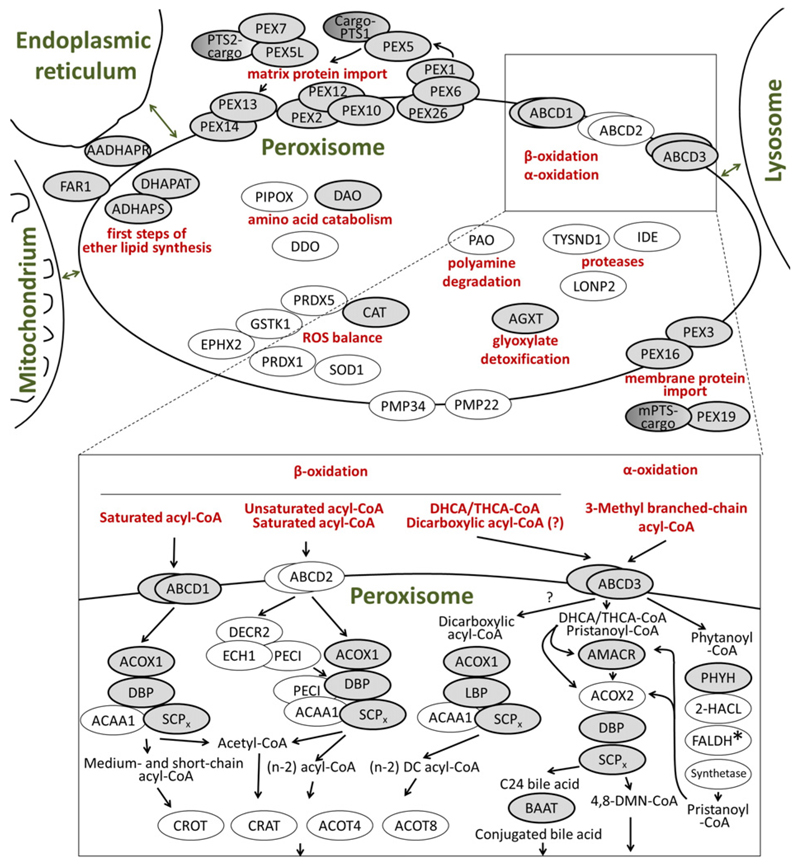
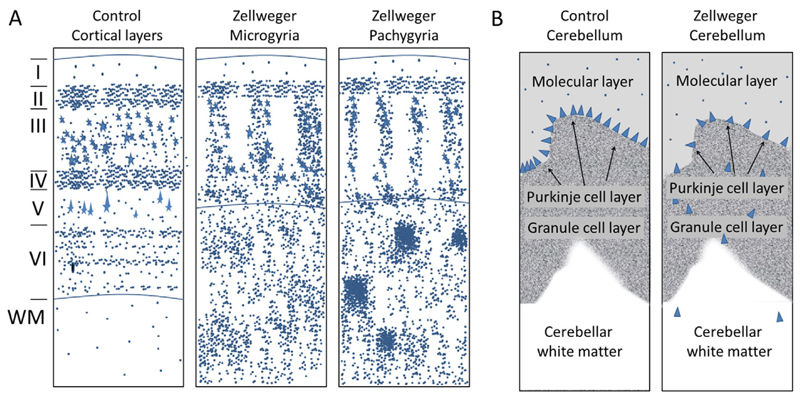
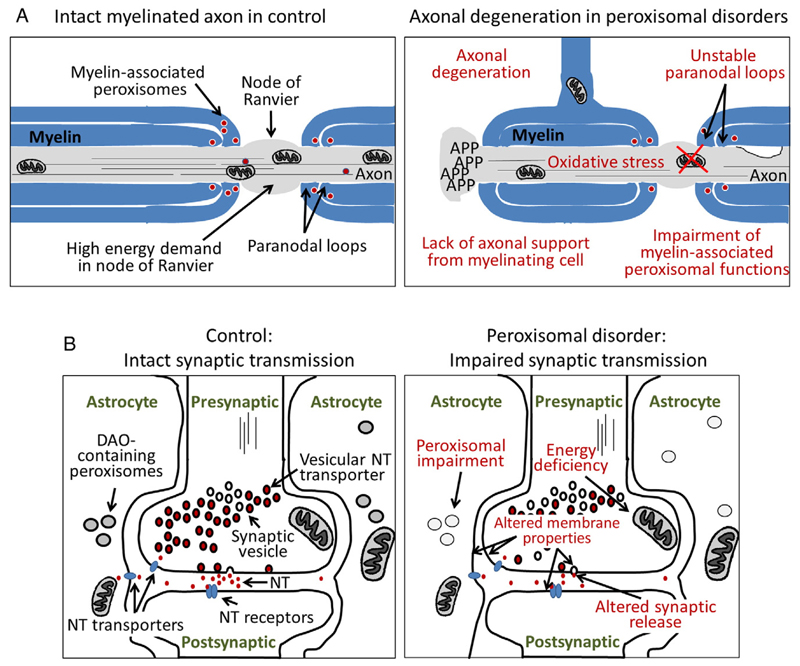
References
-
- Hruban Z, Rechcigl M., Jr Microbodies and related particles. Morphology, biochemistry, and physiology. Int Rev Cytol. 1969;(Suppl. 1):1–296. - PubMed
-
- Wiese S, Gronemeyer T, Ofman R, Kunze M, Grou CP, Almeida JA, Eisenacher M, Stephan C, Hayen H, Schollenberger L, Korosec T, et al. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol Cell Proteomics. 2007;6:2045–2057. - PubMed
-
- Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. - PubMed
-
- Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012;1822:1430–1441. - PubMed
-
- Mi J, Kirchner E, Cristobal S. Quantitative proteomic comparison of mouse peroxisomes from liver and kidney. Proteomics. 2007;7:1916–1928. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
