

Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis
- PMID: 26686880
- PMCID: PMC4703848
- DOI: 10.1038/ncomms10063
Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis
Erratum in
-
Corrigendum: Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis.Nat Commun. 2016 Apr 20;7:11465. doi: 10.1038/ncomms11465. Nat Commun. 2016. PMID: 27095245 Free PMC article. No abstract available.
Abstract
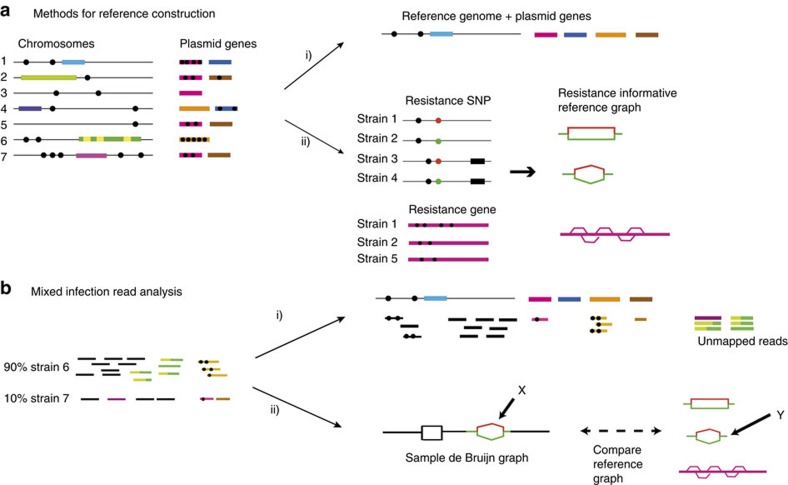
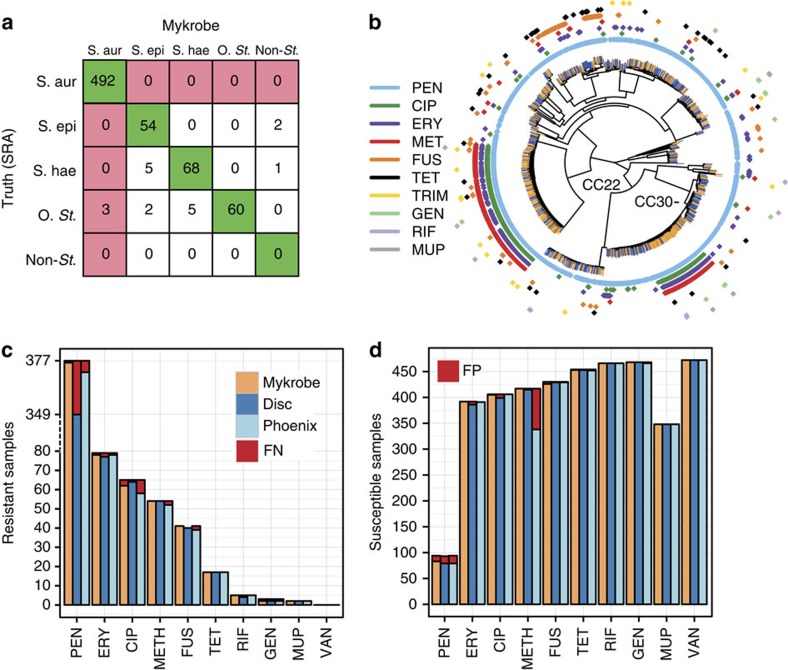
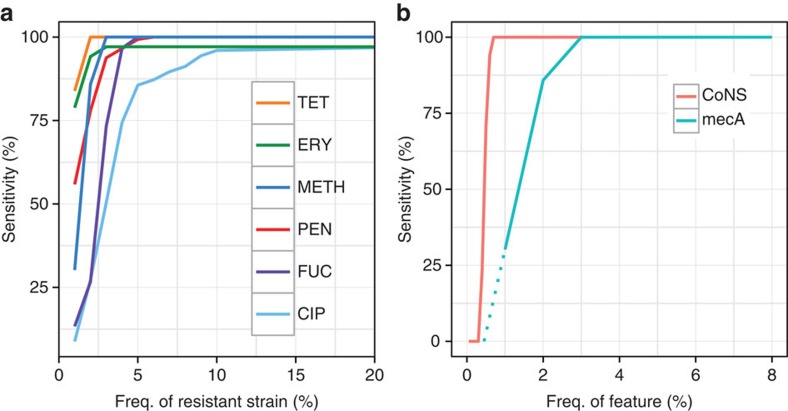
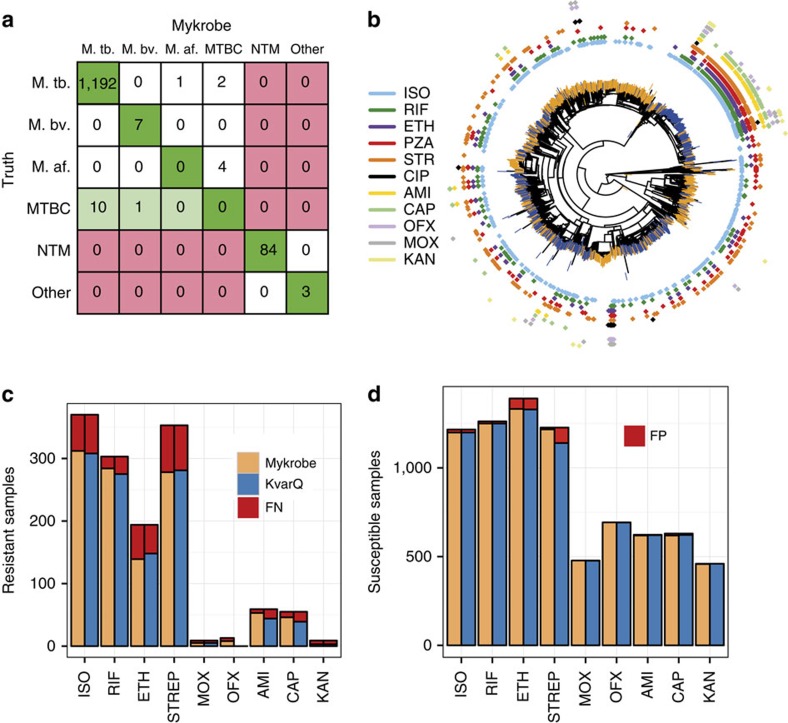
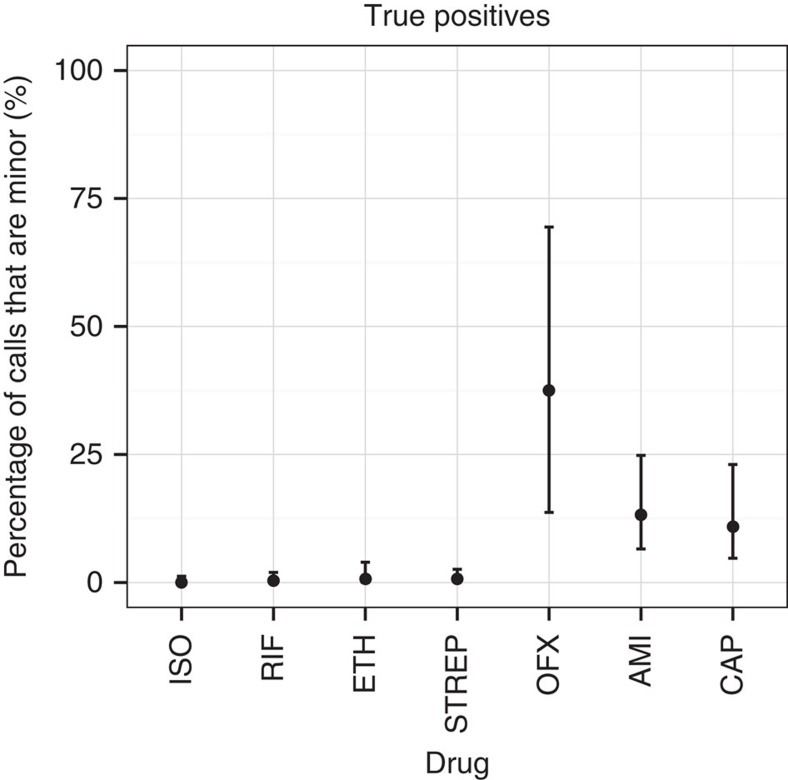
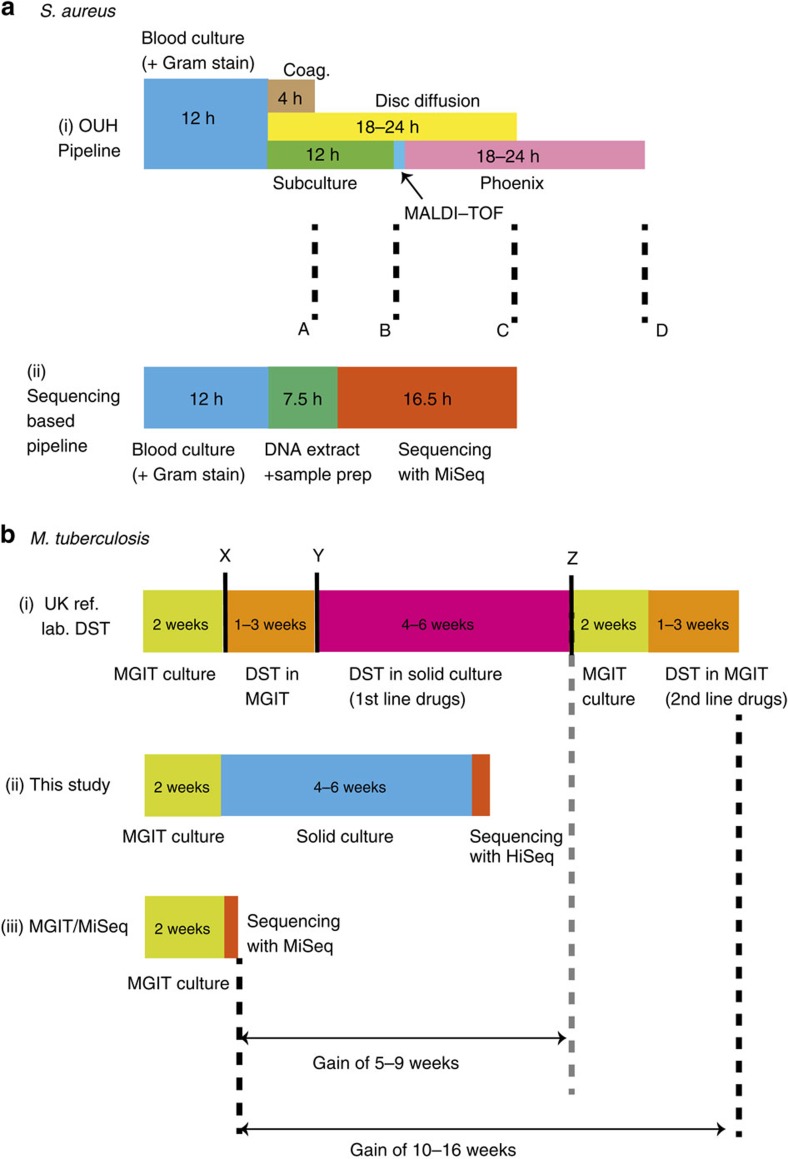
The rise of antibiotic-resistant bacteria has led to an urgent need for rapid detection of drug resistance in clinical samples, and improvements in global surveillance. Here we show how de Bruijn graph representation of bacterial diversity can be used to identify species and resistance profiles of clinical isolates. We implement this method for Staphylococcus aureus and Mycobacterium tuberculosis in a software package ('Mykrobe predictor') that takes raw sequence data as input, and generates a clinician-friendly report within 3 minutes on a laptop. For S. aureus, the error rates of our method are comparable to gold-standard phenotypic methods, with sensitivity/specificity of 99.1%/99.6% across 12 antibiotics (using an independent validation set, n=470). For M. tuberculosis, our method predicts resistance with sensitivity/specificity of 82.6%/98.5% (independent validation set, n=1,609); sensitivity is lower here, probably because of limited understanding of the underlying genetic mechanisms. We give evidence that minor alleles improve detection of extremely drug-resistant strains, and demonstrate feasibility of the use of emerging single-molecule nanopore sequencing techniques for these purposes.
Conflict of interest statement
Z.I. and P.B. are potential beneficiaries of licensing of Mykrobe predictor by the University of Oxford. The remaining authors declare no conflict of interest.
Figures
References
-
- Nathan C. & Cars O. Antibiotic resistance—problems, progress, and prospects. N. Engl. J. Med. 371, 1761–1763 (2014) . - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
