

Innate Immunity and Inflammation Post-Stroke: An α7-Nicotinic Agonist Perspective
- PMID: 26690125
- PMCID: PMC4691088
- DOI: 10.3390/ijms161226141
Innate Immunity and Inflammation Post-Stroke: An α7-Nicotinic Agonist Perspective
Abstract
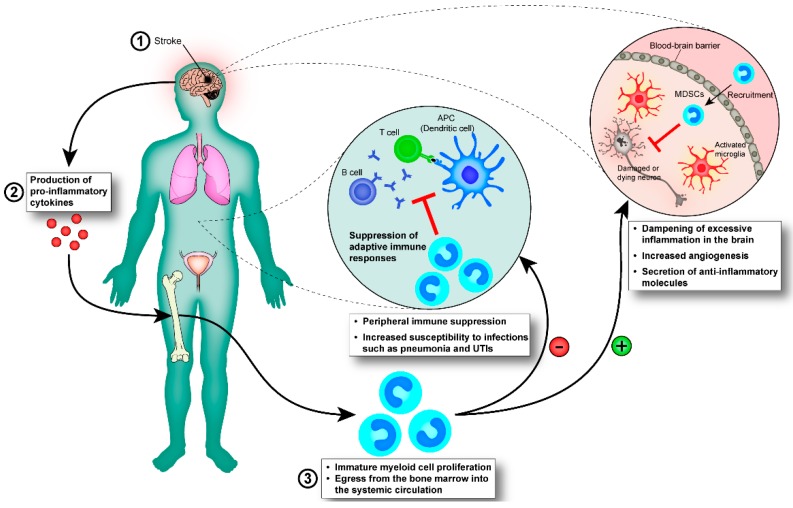
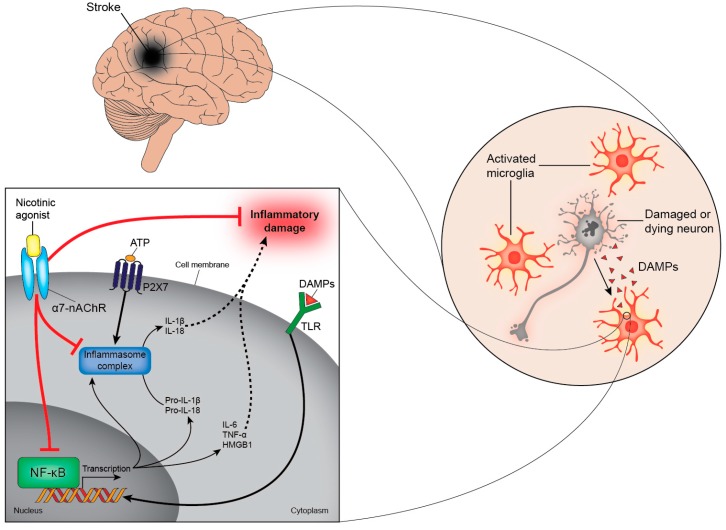
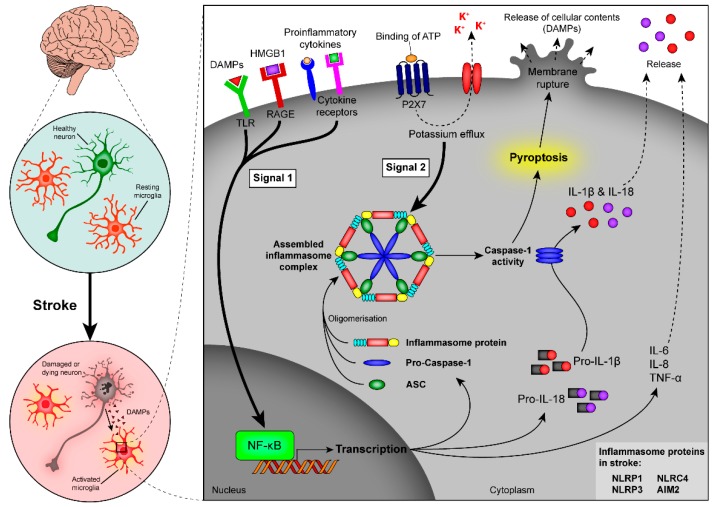
Stroke is one of the leading causes of death and long-term disability, with limited treatment options available. Inflammation contributes to damage tissue in the central nervous system across a broad range of neuropathologies, including Alzheimer's disease, pain, Schizophrenia, and stroke. While the immune system plays an important role in contributing to brain damage produced by ischemia, the damaged brain, in turn, can exert a powerful immune-suppressive effect that promotes infections and threatens the survival of stroke patients. Recently the cholinergic anti-inflammatory pathway, in particular its modulation using α7-nicotinic acetylcholine receptor (α7-nAChR) ligands, has shown potential as a strategy to dampen the inflammatory response and facilitate functional recovery in stroke patients. Here we discuss the current literature on stroke-induced inflammation and the effects of α7-nAChR modulators on innate immune cells.
Keywords: immune response; inflammation; myeloid cells; nicotinic; nicotinic acetylcholine receptor agonist; stroke.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
