

Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations
- PMID: 26691984
- PMCID: PMC4731297
- DOI: 10.1038/ng.3471
Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations
Abstract
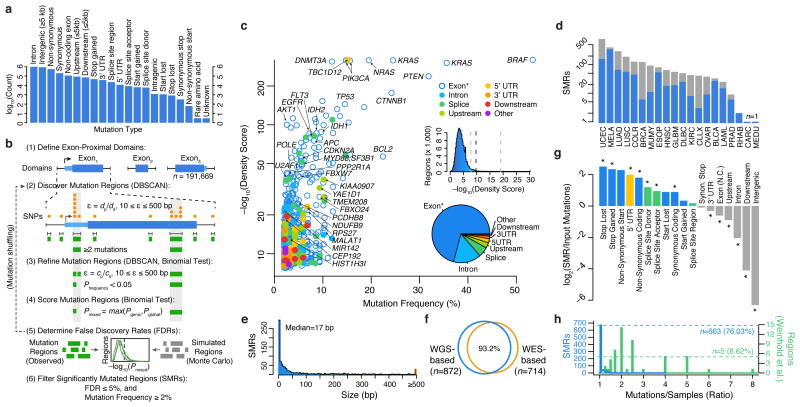
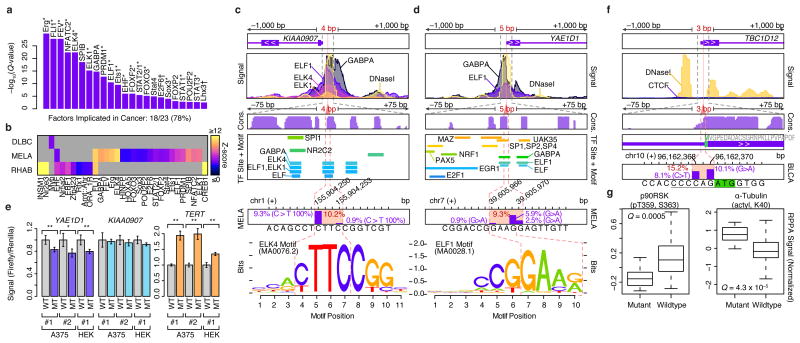
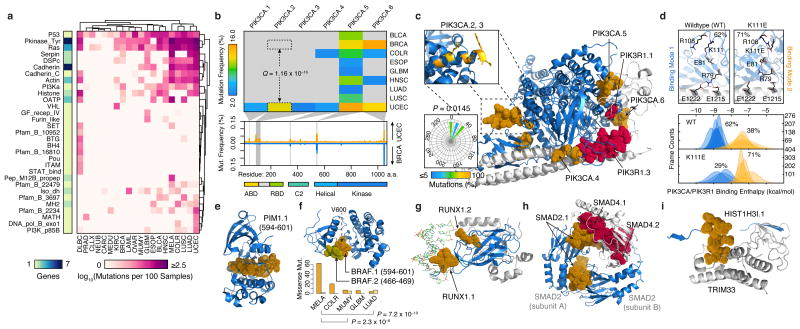
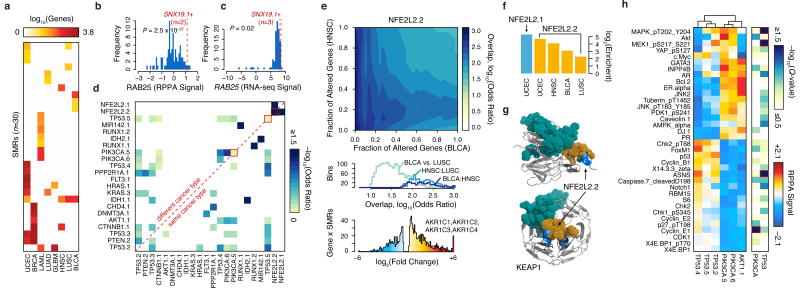
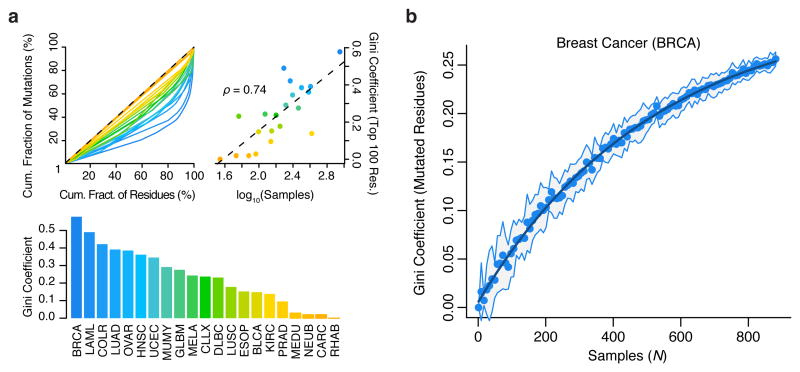
Cancer sequencing studies have primarily identified cancer driver genes by the accumulation of protein-altering mutations. An improved method would be annotation independent, sensitive to unknown distributions of functions within proteins and inclusive of noncoding drivers. We employed density-based clustering methods in 21 tumor types to detect variably sized significantly mutated regions (SMRs). SMRs reveal recurrent alterations across a spectrum of coding and noncoding elements, including transcription factor binding sites and untranslated regions mutated in up to ∼ 15% of specific tumor types. SMRs demonstrate spatial clustering of alterations in molecular domains and at interfaces, often with associated changes in signaling. Mutation frequencies in SMRs demonstrate that distinct protein regions are differentially mutated across tumor types, as exemplified by a linker region of PIK3CA in which biophysical simulations suggest that mutations affect regulatory interactions. The functional diversity of SMRs underscores both the varied mechanisms of oncogenic misregulation and the advantage of functionally agnostic driver identification.
Figures
References
Publication types
MeSH terms
Grants and funding
- U54 HG006996/HG/NHGRI NIH HHS/United States
- UL1TR000093/TR/NCATS NIH HHS/United States
- 3U54DK10255602/DK/NIDDK NIH HHS/United States
- UL1 TR000093/TR/NCATS NIH HHS/United States
- U54GM072970/GM/NIGMS NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
- P41 GM103712/GM/NIGMS NIH HHS/United States
- UL1 TR001085/TR/NCATS NIH HHS/United States
- P41GM103712-S1/GM/NIGMS NIH HHS/United States
- 1P50HG00773501/HG/NHGRI NIH HHS/United States
- U54 DK102556/DK/NIDDK NIH HHS/United States
- 1U01HG007919-01/HG/NHGRI NIH HHS/United States
- U01 HG007919/HG/NHGRI NIH HHS/United States
- U54 GM072970/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
