

A significant risk locus on 19q13 for bipolar disorder identified using a combined genome-wide linkage and copy number variation analysis
- PMID: 26692414
- PMCID: PMC4683747
- DOI: 10.1186/s13040-015-0076-y
A significant risk locus on 19q13 for bipolar disorder identified using a combined genome-wide linkage and copy number variation analysis
Abstract
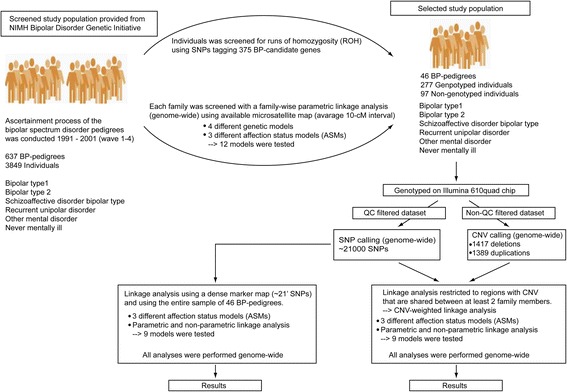
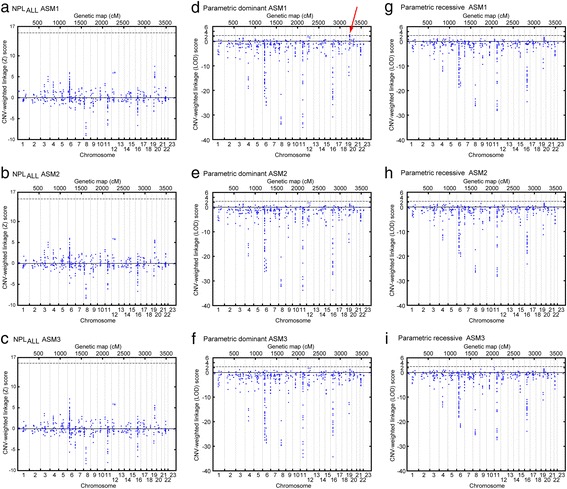
Background: The genetic background to bipolar disorder (BPD) has been attributed to different genetic and genomic risk factors. In the present study we hypothesized that inherited copy number variations (CNVs) contribute to susceptibility of BPD. We screened 637 BP-pedigrees from the NIMH Genetic Initiative and gave priority to 46 pedigrees. In this subsample we performed parametric and non-parametric genome-wide linkage analyses using ~21,000 SNP-markers. We developed an algorithm to test for linkage restricted to regions with CNVs that are shared within and across families.
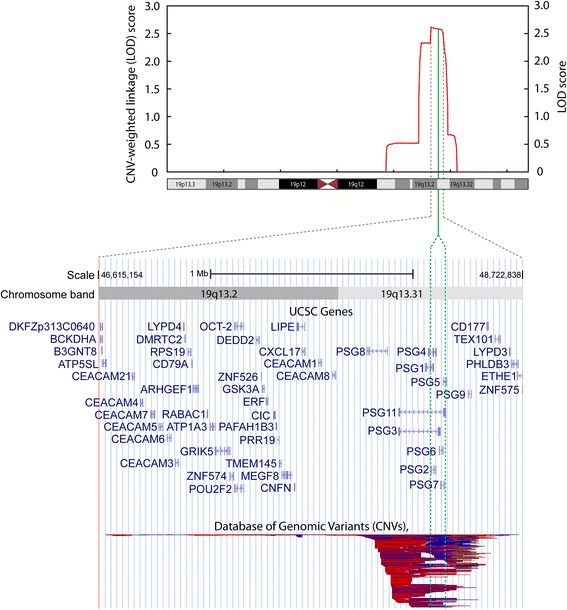
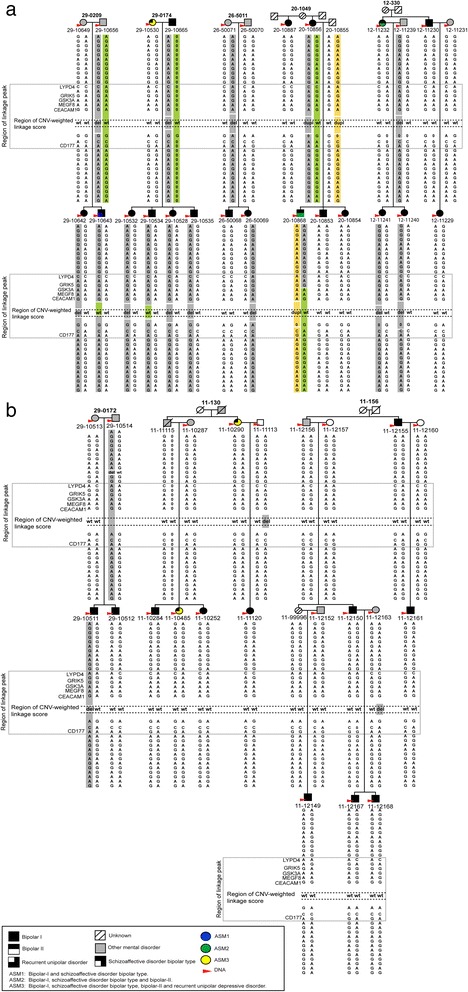
Results: For the combined CNV and linkage analysis, one region on 19q13 survived correction for multiple comparisons and replicates a previous BPD risk locus. The shared CNV map to the pregnancy-specific glycoprotein (PSG) gene, a gene-family not previously implicated in BPD etiology. Two SNPs in the shared CNV are likely transcription factor binding sites and are linked to expression of an F-box binding gene, a key regulator of neuronal pathways suggested to be involved in BPD etiology.
Conclusions: Our CNV-weighted linkage approach identifies a risk locus for BPD on 19q13 and forms a useful tool to future studies to unravel part of the genetic vulnerability to BPD.
Keywords: Bipolar disorder; Copy number variation; Genome-wide; Linkage analysis.
Figures
Similar articles
-
Genome-wide association study of copy number variation with lung function identifies a novel signal of association near BANP for forced vital capacity.BMC Genet. 2016 Aug 11;17(1):116. doi: 10.1186/s12863-016-0423-0. BMC Genet. 2016. PMID: 27514831 Free PMC article.
-
Association study of 83 candidate genes for bipolar disorder in chromosome 6q selected using an evidence-based prioritization algorithm.Am J Med Genet B Neuropsychiatr Genet. 2013 Dec;162B(8):898-906. doi: 10.1002/ajmg.b.32200. Epub 2013 Sep 30. Am J Med Genet B Neuropsychiatr Genet. 2013. PMID: 24123842
-
Genome-wide copy number variation-, validation- and screening study implicates a new copy number polymorphism associated with suicide attempts in major depressive disorder.Gene. 2020 Sep 10;755:144901. doi: 10.1016/j.gene.2020.144901. Epub 2020 Jun 15. Gene. 2020. PMID: 32554045
-
Review of bipolar molecular linkage and association studies.Curr Psychiatry Rep. 2002 Apr;4(2):124-9. doi: 10.1007/s11920-002-0045-2. Curr Psychiatry Rep. 2002. PMID: 11914173 Review.
-
Searching high and low: a review of the genetics of bipolar disorder.Bipolar Disord. 2000 Mar;2(1):8-26. doi: 10.1034/j.1399-5618.2000.020103.x. Bipolar Disord. 2000. PMID: 11254025 Review.
References
-
- Hirschfeld RM, Vornik LA. Bipolar disorder--costs and comorbidity. Am J Manag Care. 2005;11:S85–90. - PubMed
-
- Marneros A, Angst J. Bipolar Disorders, 100 years after manic-depressive insanity. New York: Kluwer Academic Publishers; 2002.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
